目录
生信领域大神李恒今年发在预印本上的一篇综述:Genome assembly in the telomere-to-telomere era,小编总结下要点。
影响组装的基因组特性
决定基因组组装难易程度的主要因素不是基因组大小,而是它的重复结构。重复序列可以通过比其更长的reads来解析。但是,还有更长的重复区域。例如,人类 1 号染色体的着丝粒周围区域含有 20 Mb 的重复序列比当前测序技术产生的读长要长得多。但我们仍然可以通过准确的长读长来组装这个区域。尽管它和其他此类区域由相似的重复拷贝组成,但它们随着时间的推移积累了突变,并且很少在10 kb以上共享相同的重复序列。给定长的无错误读长,我们可以区分不同的重复拷贝并成功组装它们。Reads从来都不是完全没有错误的,但是当reads错误率足够低并且测序错误足够独立时,我们可以纠正大多数错误并实现高质量的组装。
重复序列大致可分为三类:间隔重复序列(interspersed repeats)、串联重复序列(tandem repeats)和片段重复序列(segmental duplications)。
- 间隔重复序列大多是分散在基因组中的转座元件。它们几乎都比现代长读长读短,因此不再对组装构成重大挑战。
- 染色体臂上的大多数串联重复序列比长读长序列短,因此也易于组装。然而,卫星重复序列(satellite repeats)是一种通常富含着丝粒的超长串联重复序列,特别难以组装,因为整个卫星阵列不能被长读长跨越。
- 片段重复是指基因组中重复的非常长的DNA片段,通常比长reads甚至超长reads要长。它们中的许多是聚集在一起的,可以串联。虽然古老的固定区段重复很容易解决,因为它们自其共同祖先以来通过突变积累了差异,但长多态性重复具有挑战性。
- 核糖体 DNA (rDNA) 可以组织为由高度相似的拷贝组成的长串联阵列。长 rDNA 阵列是最难组装的区域之一。
长读长和长范围(long-range)测序技术
长读长技术可生成长度通常为 ≥10 kb 的连续读序列。
2019 年,PacBio 推出了长度为 10–20 kb 且错误率低于 0.5% 的高保真 (HiFi) 读长。这些有效地取代了 PacBio 错误率为 >10% 的旧连续长读长 (CLR)。
目前面向大众市场的 ONT 产品的准确率大致在 90-95% 左右,长度为 ≥100 kb。最新的 ONT v14 化学反应可以使用最新的 Dorado 碱基调用器生成准确率为 98-99% 的读数。ONT正在积极开发双链测序技术,该技术可对DNA片段的两条链进行测序,在准确性上接近 PacBio HiFi,并且可以更长。
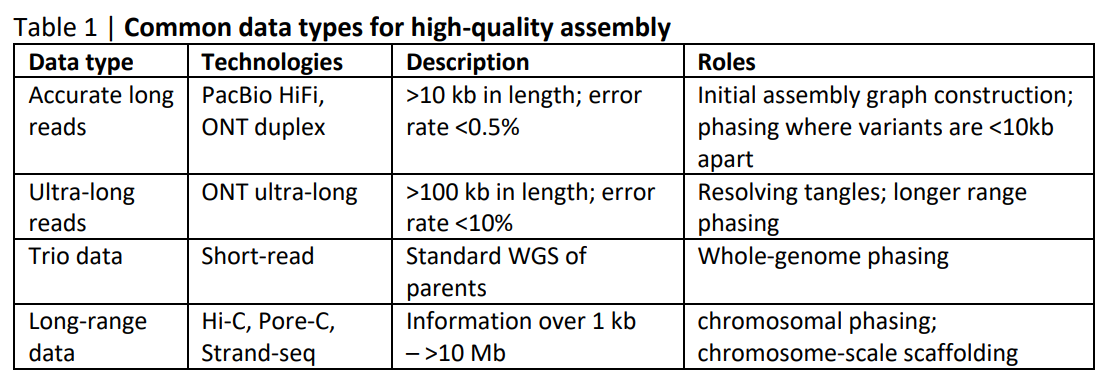
即使是超长读长,也很少跨越超过几百Kb。为了可靠地获得染色体长支架和相位,需要长范围数据,使用最广泛的是Hi-C。Pore-C 与 Hi-C 类似,但使用 ONT 测序。Strand-seq 是另一种特别擅长染色体分组和重叠群定向的技术,但更昂贵,并且无法在市场上买到。亲本序列数据或三重数据(trio data)对于全基因组定相非常强大,也可以被视为一种长范围数据。
近T2T基因组组装
对于纯合基因组,近T2T组装的最可靠解决方案同时使用 PacBio HiFi reads 和 ONT 超长读长。一般先用 HiFi reads 来构建一个初始组装图,再用超长填补间隙。
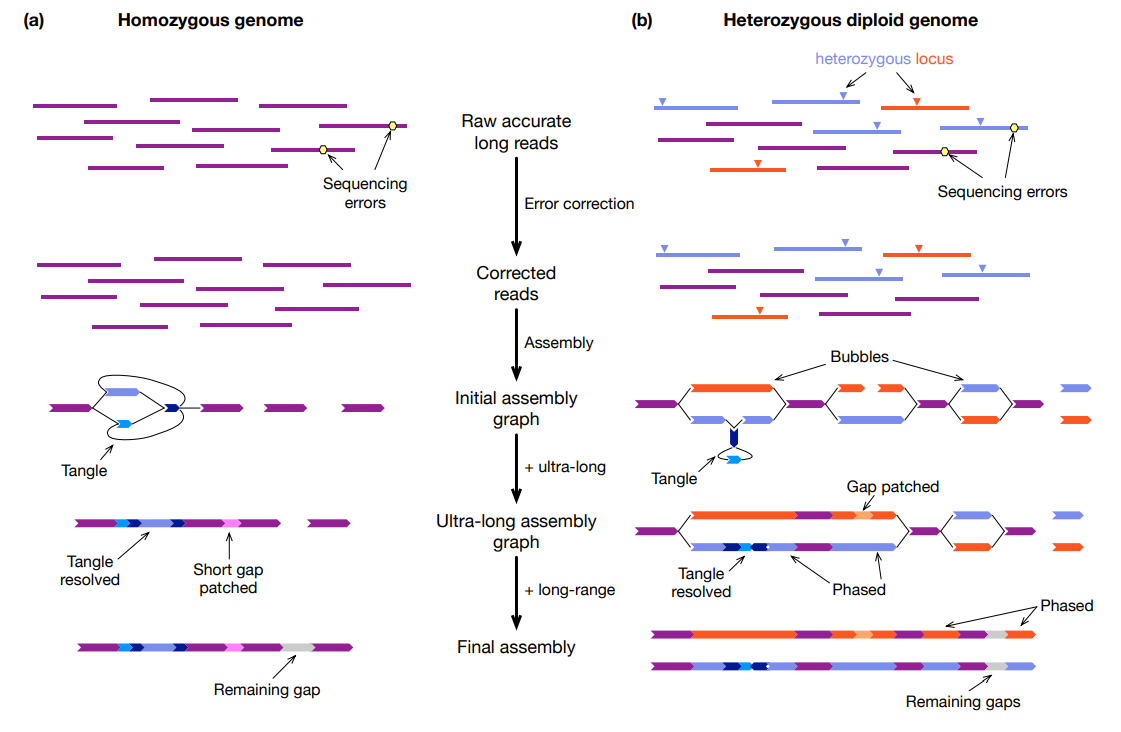
组装杂合二倍体基因组遵循类似的策略。对于具有长纯合区域的基因组,单独使用HiFi和超长的组合可能无法使整个染色体分相。在这种情况下,建议使用三重数据来提供整个基因组的准确分相。当无法获得亲本样本时,可能使用Hi-C代替。Hi-C 仅提供重叠群之间的相对相位信息,不如三重数据强大,尤其是在纠结的子图中,但Hi-C仍然是可靠支架染色体的关键数据类型。将 HiFi 与 trio、Hi-C 或 Strand-seq 等长范围数据相结合,可以产生一对单倍型分辨组装,此组装具有相当的连续性。它还保留了相位,并且可以进一步用 Hi-C 搭建成分相染色体。
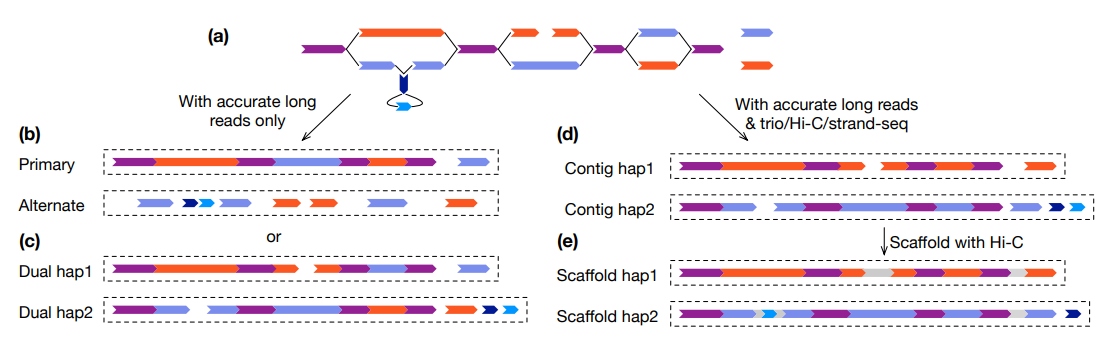
核心组装算法
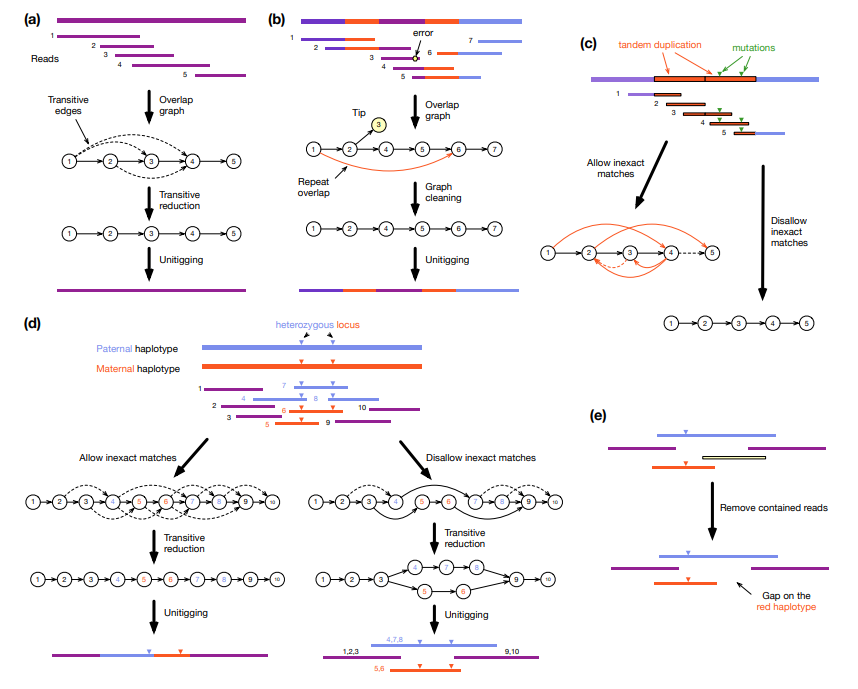
现代长读长组装基本都是基于图论,即overlap graph或de Bruijn graph。在此图中,顶点表示一个序列,一条边表示从读取中推断出的可能连接。理想情况下,组装图将所有信息保留在读取中,没有冗余。然而,由于重复和倍性,它通常是非线性的。
评估序列组装
基本指标
组装大小、contig长度综合、N50等。对于二倍体常染色体组装要有一对分相组装且具有相似大小,一对不平衡的常染色体组装可能表明分相不完整,可能要手动参数调整。性染色体很可能具有不同的大小。物种内的其他倍性变异也可能发生,例如由于体细胞染色体丢失或减少。
评估基因完整性
BUSCO是首选。minimap2 软件包中的“asmgene”工具是 BUSCO 的替代方案,还可以解决存在高质量参考基因组时的低完整性问题。
基于 K-mer 的评估
假设 k-mer 的计数与其读长计数成正比, k-mer 在读长中具有高频率但组装中不存在,表明序列缺失。KAT 是一个强大的工具,它利用这些简单的观察结果来评估组装。
使用k-mers来估计重叠群序列的基本准确度是一种常见的做法,通常以Phred scake作为QV(Quality Value)进行测量。目前有两种实现,Merqury 和 yak 。
基于比对的评估
理想情况下,当我们将序列读长与其组装比对时,我们期望在每个重叠群位置都能均匀覆盖。在较长的区域上覆盖率过低或过高都表明存在潜在的组装错误。我们还希望重叠群能够得到基础级别的读取的良好支持。Flagger、Asset和 Inspector是基于读长到程序集比对的面向用户的评估工具。
对于具有近乎完美的基因组,可以此作为基本事实,以评估使用较少数据类型或较低读取覆盖率生成的自动化程序集。QUAST 是很好的工具,这种基于比对的方法对于开发人员调整组装算法非常宝贵,但不适用于新物种,或者当“真实”组装和评估组装来自不同的菌株或不同样品时。
李恒的观点
在讨论部分,李恒指出了overlap-based和DBG两种算法组装、HiC数据以及组装软件的不足,他很看好ONT最新的simplex reads,兼顾超长与准确性,可能会大大简化高质量基因组组装。
我们能否用当前数据自动组装从端粒到端粒的所有染色体?李恒认为是不行的。他认为,过去几年的大部分进步都是由于数据质量的提高而取得的,而当前的软件从可用的输入数据中提取了大部分信息。仅靠算法改进可能无法可靠地解决所有组装gap。
我们期待在测序技术方面不断取得新的进展,以便在没有人为干预的情况下真正完成基因组。需要注意的是,一个完整的组装只是为下游生物学发现设定了一个开始。虽然基因组组装进展迅速,但基因组比对和注释工具却远远落后。我们希望在未来看到这些工具的持续发展,以实现(近乎)完整组装的全部功能。
更多信息请关注:
