多基因组比对 (multiple genome alignment, MGA)首先要定义多序列比对 (multiple sequence alignment, MSA)。MSA 是将同源关系分配给 3 个或更多序列的方法(对于 2 个序列,使用“成对”而非“多个”),其中一组核苷酸是同源的,如果它们来自同一个共同祖先。这些比对通常由二维数组表示,其中每行代表一个输入序列,每列代表一组同源核苷酸。
MSA 具有严格的约束,对齐是共线的,即从左到右读取的每一行,忽略空列,必须是原始输入序列。因此,MSA只能捕获小的插入缺失和点突变。找到最大化同源性的比对是一项计算困难的任务。此外,MSA不模拟进化事件,如倒位、易位和整个基因的获得或损失。在某些情况下,这些序列在医学上与人类疾病有关。尽管存在缺点,但MSA是研究同源关系的关键第一步,是准确重建系统发育树的重要前提,它也是有史以来研究最多的科学问题之一。
MGA的一个核心问题是如何正确地找到同源区域。由于 MGA 包括寻找和比对适合 MSA 的输入基因组的同源区域,因此 MGA 本质上与 MSA 一样具有挑战性。
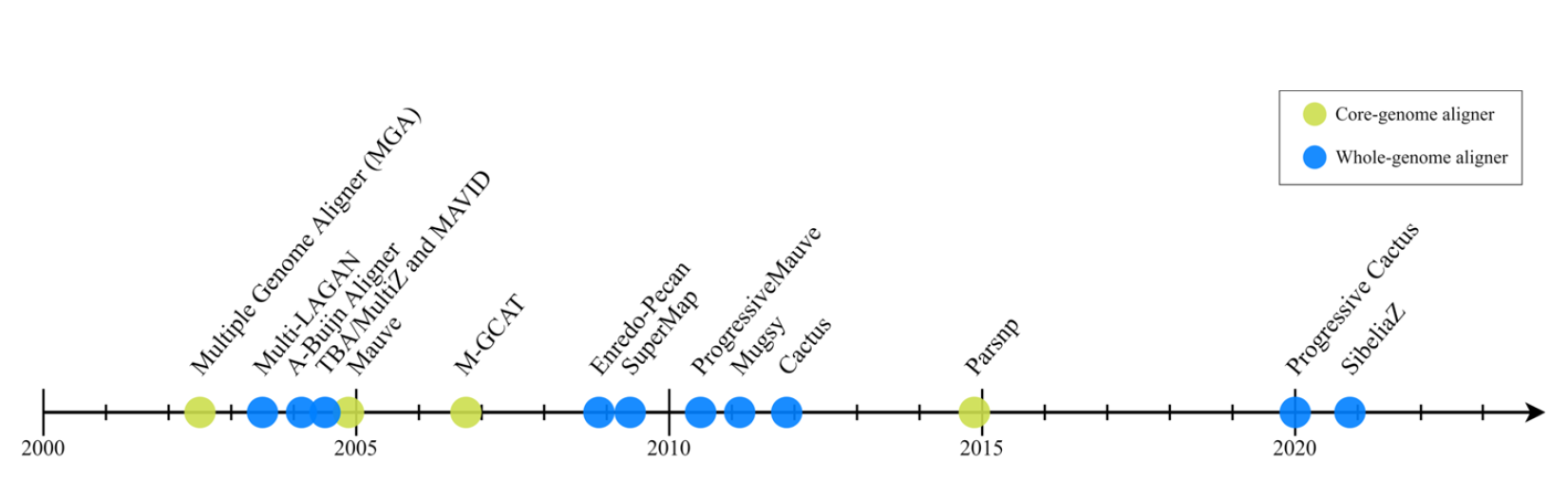
由于全基因组比用于MSA的序列更长,结构更多样化,因此几乎所有MGA算法都使用两步程序来分解问题:首先在两个或多个序列中识别高度相似的区域,称为锚点,然后使用这些锚点在输入基因组中识别更大的无重排区域,称为局部共线块(locally colinear blocks, LCB)。
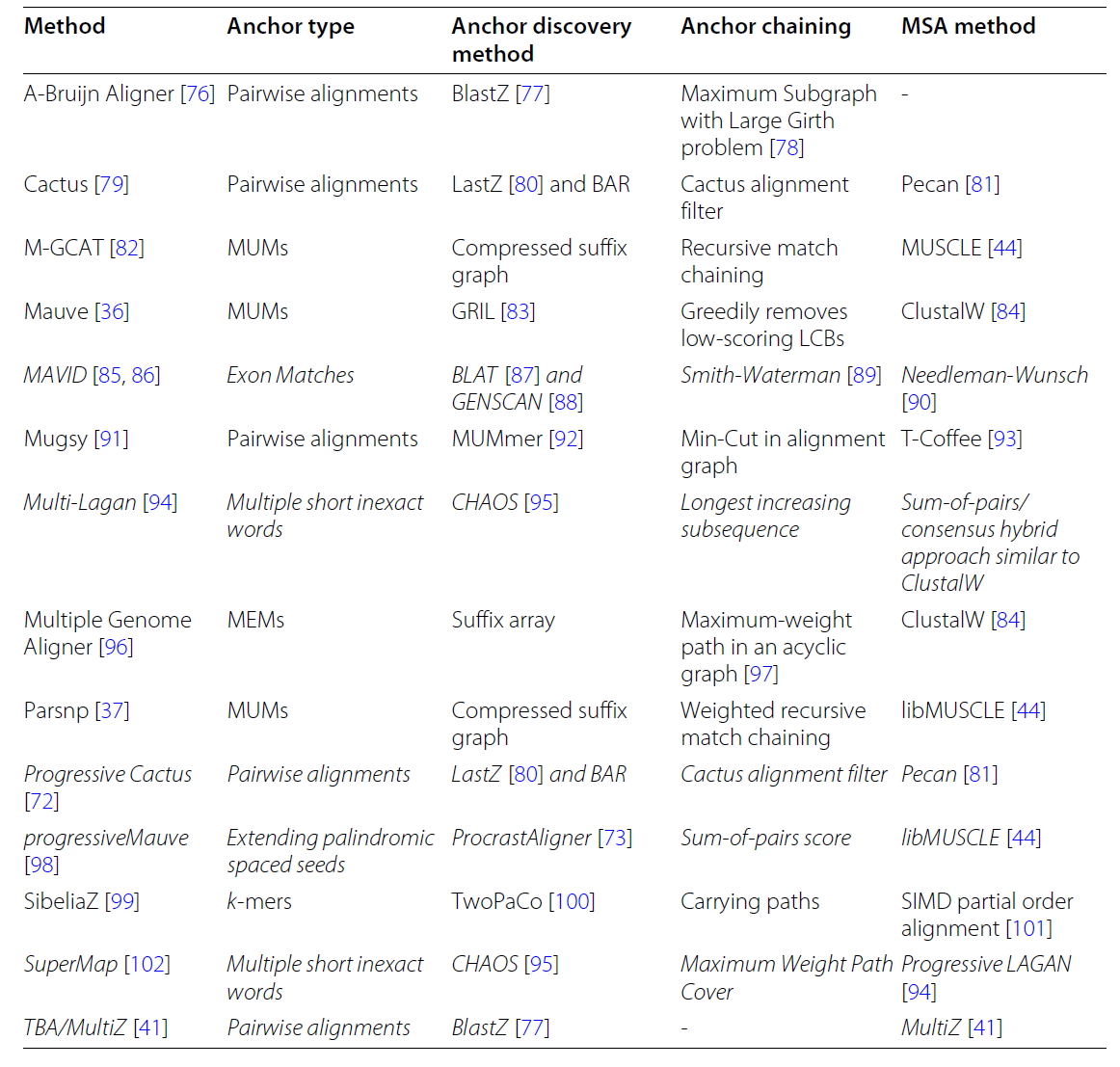
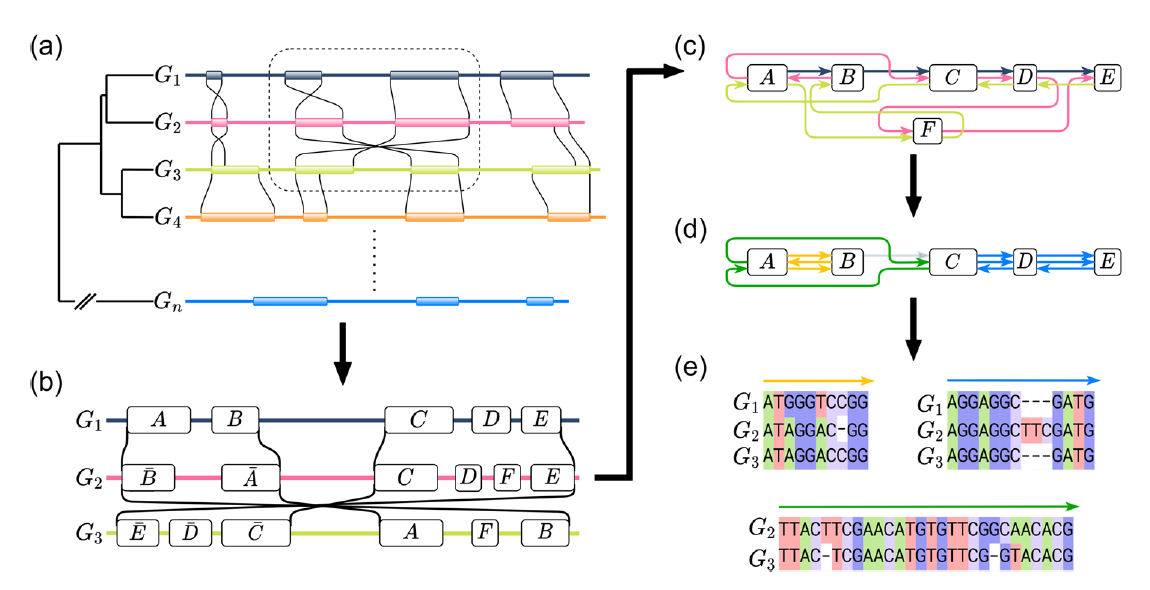
锚点算法:
- 成对精确:MUMmer
- 成对近似值:LastZ
- 多序列精确:Parsnp
- 多序列近似值:ProcrastAligner
对齐算法:
- Basic alignment graphs
- A-Bruijn graphs
- Enredo graphs
- Cactus graphs
- de-Bruijn graphs
LCB构建:
- Graph-free LCB construction: ProgressiveMauve
- A-Bruijn graphs: Mugsy
- Enredo graphs: Enredo-Pecan aligner
- Cactus graphs: Cactus aligner
- De-Bruijn graphs: SibeliaZ
随着基因组的完成和重复区域的正确解析,MGA算法可以更加专注于查找准确的局部比对,减少调整测序误差的需要。由于多基因组比对是一个广泛的领域,未来的改进可能包括重复屏蔽、局部比对、图论、LCB构建和多序列比对等方向。
Kille B, Balaji A, Sedlazeck FJ, Nute M, Treangen TJ. Multiple genome alignment in the telomere-to-telomere assembly era. Genome Biol. 2022;23(1):182. Published 2022 Aug 29. doi:10.1186/s13059-022-02735-6
更多信息请关注微信公众号:生物信息与育种
