目录
本文整理自Giovanny E Covarrubias Pazaran博士的报告《Genomic Selection in R》,他是国际水稻研究所植物育种和遗传学博士,具有强大的统计学背景以及具有从事育种计划和实践研究的丰富经验,是R和SAS等不同软件平台进行多元线性混合建模的专家,开发了著名的多变量基因组选择混合模型R包软件sommer。
在这里,Giovanny基于sommer总结了GS应用的四种应用场景,对于知道GS但不了解GS如何应用的朋友可能有所帮助。
大多数现有的基因组选择软件只能计算包含一个方差组分,除了误差之外,这使得对具有杂交效应的物种使用加性、显性和上位效应进行杂种预测不可行。R包sommer促进了使用混合模型进行基因组选择和杂交预测,使用多个方差组分并允许指定协方差结构。包括四种用于估计方差组分的算法:平均信息(AI)、牛顿-拉弗逊(NR)、期望最大化(EM)和高效混合模型关联(EMMA;岭回归)。包括用于计算加性、显性和上位关系矩阵的核函数,以及用于基因组分析的其他有用函数。
sommer可以处理比常规基因组选择软件更复杂的问题,比贝叶斯更快,时间在小时到几天的数量级,可以处理缺失数据,使用R等友好的环境。其他可用于基因组选择的软件包括:
- rrBLUP (仅适用于单一随机效应)
- regress (只有 NR 算法可用,返回负方差组分)
- ASReml (收费)
- BGLR (贝叶斯算法可能需要很长时间)
- MCMCglmm (更好的贝叶斯方法,但需要很长时间)
GS应用的四个场景
预测杂交种的总体表现
(1)亲本的基因型和表型数据可用,希望根据纯加性模型(没有杂种优势的物种)来预测可能杂交种的表现。
(2)亲本的基因型数据可用,某些可能的杂交组合有表型数据可用(约10%),希望根据加性-显性模型(具有杂种优势的物种)来预测其余可能杂交种的表现(约90%)。
预测群体内个体的具体表现
(3)群体个体的基因型数据可用,但只有某些个体有表型数据(即表型测定非常昂贵),目标是使用纯加性模型来预测其余个体的表现。
(4)群体个体的基因型数据可用,但只有某些个体有表型数据(即表型测定非常昂贵),目标是使用加性-显性-上位基因模型来预测其余个体的表现。
情景1
处理繁殖方式是自交的物种,杂种优势较少出现。在这种情况下,杂交的表现可以估算为亲本育种值(BV)的平均值。为了获得特定亲本的育种值,这些材料会在不同的地点和年份进行测试,并拟合混合模型以获得基因型的BLUP值。
Henderson认为,当一些数据缺失时,可以利用个体之间的系谱关系来预测在数据缺失时一些个体的表现。跟踪系谱在所有育种计划中都很困难,这导致了使用标记来估算亲缘关系的出现。基于标记的这种关系矩阵被命名为基因组关系矩阵,与基于家系的加性关系矩阵类似。
假设您在CIMMYT工作,对599份小麦材料的1279个SNP标记的基因组信息进行了收集。鉴于这些材料是线性的,预计只有加性方差是显著的。现在,若希望预测这599份材料之间的所有可能杂交的表现,使用sommerR包,可以这样做。
#### call the phenotypic and genotypic information
library(sommer)
data(wheatLines)
X <- wheatLines$wheatGeno; X[1:5,1:5]; dim(X)
Y <- wheatLines$wheatPheno
rownames(X) <- rownames(Y)
#### select environment 1 and create incidence and additive
#### relationship matrices
y <- Y[,1] # response grain yield
Z1 <- diag(length(y)) # incidence matrix
K <- A.mat(X) # additive relationship matrix
#### perform the GBLUP pedigree-based approach by
### specifying your random effects (ETA) in a 2-level list
### structure and run it using the mmer function
ETA <- list(add=list(Z=Z1, K=K))
ans <- mmer(y=y, Z=ETA, method="EMMA") # kinship based
summary(ans)
#### Predict the progeny by extracting the BV for the lines
#### and get the average BV for all possible combinations
GEBV.pb <- ans$u.hat # this are the BV
rownames(GEBV.pb) <- rownames(Y)
crosses <- do.call(expand.grid, list(rownames(Y),rownames(Y)));
dim(crosses)
cross2 <- duplicated(t(apply(crosses, 1, sort)))
crosses2 <- crosses[cross2,]; head(crosses2); dim(crosses2)
# get GCA1 and GCA2 of each hybrid
GCA1 = GEBV.pb[match(crosses2[,1], rownames(GEBV.pb))]
GCA2 = GEBV.pb[match(crosses2[,2], rownames(GEBV.pb))]
#### join everything and get the mean BV for each combination
BV <- data.frame(crosses2,GCA1,GCA2); head(BV)
BV$BVcross <- apply(BV[,c(3:4)],1,mean); head(BV)
plot(BV$BVcross)
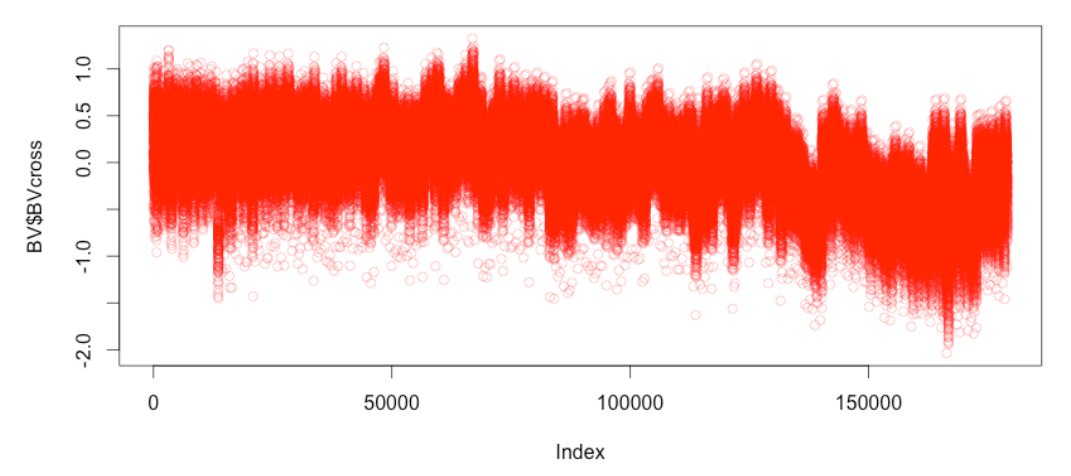
最后,您将获得这599份小麦品系之间的179,101个可能杂交的基因组估值(GEBV),可以按最佳表现对它们进行排序,在实际育种中,将选择预测中表现最佳的杂交组合。
情景2
处理繁殖方式是杂交的物种,通常会存在杂种优势。在这个例子中,展示了如何使用已有的单交信息来对尚未发生的单交种进行基因组预测。假设您在一个玉米育种计划中工作,有来自两个杂种优势群(Dent和Flint)的40个个体,每个群20个。已知这40个亲本的基因型数据,并且在四个环境中对可能的400个单交种进行了表型评估,得到了其中100个的信息。您可以使用这些信息来预测其他300个单交种的表现。
data(cornHybrid)
hybrid2 <- cornHybrid$hybrid # extract cross data
A <- cornHybrid$K # Additive relationship matrix for all
y <- hybrid2$Yield # response
### incidence matrices
X1 <- model.matrix(~ Location, data = hybrid2);dim(X1)
Z1 <- model.matrix(~ GCA1 -1, data = hybrid2);dim(Z1)
Z2 <- model.matrix(~ GCA2 -1, data = hybrid2);dim(Z2)
Z3 <- model.matrix(~ SCA -1, data = hybrid2);dim(Z3)
#### Realized IBS relationships for each effect
K1 <- A[levels(hybrid2$GCA1), levels(hybrid2$GCA1)]; dim(K1)
K2 <- A[levels(hybrid2$GCA2), levels(hybrid2$GCA2)]; dim(K2)
S <- kronecker(K1, K2) ; dim(S)
rownames(S) <- colnames(S) <- levels(hybrid2$SCA)
### specify random component
ETA <- list(list(Z=Z1, K=K1), list(Z=Z2, K=K2), list(Z=Z3, K=S))
ans <- mmer(y=y, X=X1, Z=ETA)
summary(ans)
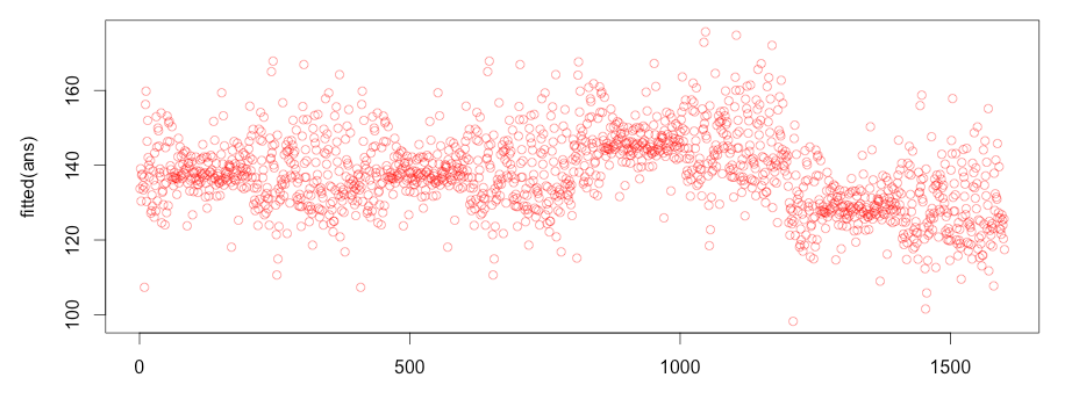
现在,您已经得到了所有可能的400个单交杂交种的拟合值,包括那些缺失的数据点,以及一般配合力(GCA)和特殊配合力(SCA)的BLUP值。

情景3
发生在想要预测特定个体的表现,已获得其基因型数据但没有表型数据。通常,这种情况发生在表型测定昂贵且只能测定其中一些个体时,但基因型测定不受限制。因此,您可以利用所有信息来预测那些已被基因型测定但没有表型数据的个体的表现。我们将使用一个纯加性模型来预测全同胞家系中个体的颜色。
data(CPdata)
CPpheno <- CPdata$pheno
CPgeno <- CPdata$geno
### look at the data
head(CPpheno)
CPgeno[1:5,1:5]
## fit a model including additive and dominance effects
y <- CPpheno$color
Za <- diag(length(y))
A <- A.mat(CPgeno) # additive relationship matrix
y.trn <- y # copy the response to test prediction accuracy
### delete data for 1/5 of the population
ww <- sample(c(1:dim(Za)[1]),72)
y.trn[ww] <- NA
ETA.A <- list(add=list(Z=Za,K=A))
ans.A <- mmer(y=y.trn, Z=ETA.A)
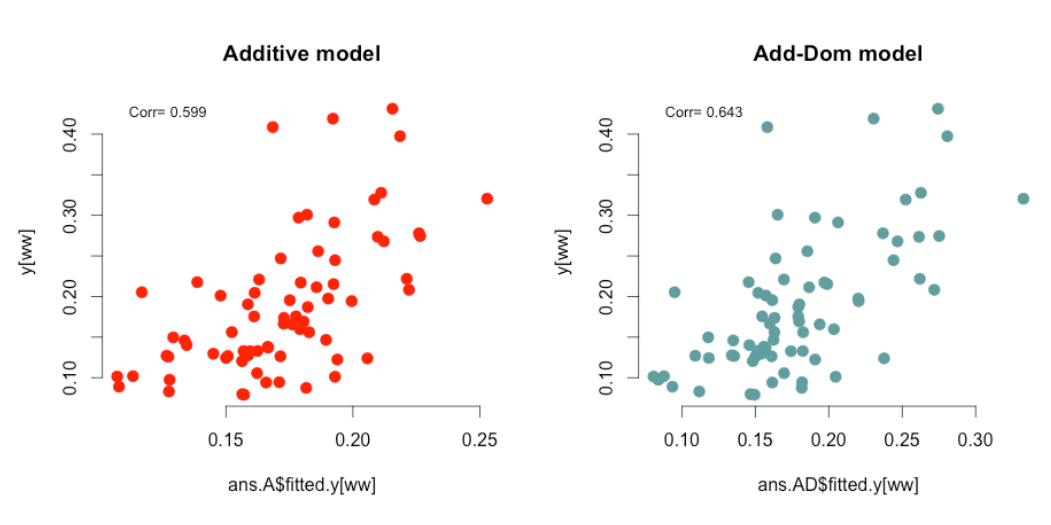
cor(ans.A$fitted.y[ww], y[ww], use="pairwise.complete.obs")
情景4
与情景3相同,但将显性和上位基因关系添加到模型中。考虑到这是一个全同胞家系,并且我们知道σ2D的四分之一是在这种类型家系的个体之间共享的,模型将包括这些效应,以期望提高预测准确性。
Zd <- diag(length(y))
Ze <- diag(length(y))
D <- D.mat(CPgeno) # dominant relationship matrix
E <- E.mat(CPgeno) # epistatic relationship matrix
ETA.ADE <- list(add=list(Z=Za,K=A),dom=list(Z=Zd,K=D),epi=list(Z=Ze,K=E))
ans.ADE <- mmer(y=y.trn, Z=ETA.ADE)
cor(ans.ADE$fitted.y[ww], y[ww], use="pairwise.complete.obs")
summary(ans.ADE)
正如您所见,上位基因方差通常为零或微不足道,不能在预测中产生差异,但加入显性关系确实提高了全同胞家系的预测准确性,这与理论一致。您可能希望时不时进行检查,并在处理这些家系或多倍体物种时使用它。
标签:基因型,场景,GS,预测,基因组,个体,表型 From: https://www.cnblogs.com/miyuanbiotech/p/17804052.htmlGiovanny作为GS领域的专家,稍了解过的同学可能知道他讲解过CGIAR Excellence in Breeding Plantform(EiB)举办的《Basics of Genomic Selection》课程,感兴趣的可以去看看小编之前整理的推文:CGIAR基因组选择合集