动脉粥样硬化(Atherosclerosis, as)是一种以动脉血管壁炎症和斑块积聚为特征的血管病变,是大多数心血管疾病的重要病因。除了脂质沉积和慢性炎症外,越来越多的证据表明表观遗传修饰与动脉粥样硬化越来越相关,并从治疗和生物标志物的角度都很有意义。本文就DNA甲基化和组蛋白翻译后修饰在动脉粥样硬化中的作用进行综述,并对今后动脉粥样硬化治疗的研究方向和潜在治疗方法进行了讨论。
背景(Background)
动脉粥样硬化是大多数心血管疾病的重要病因,包括卒中(中风)和冠状动脉疾病(冠心病),是全球范围内死亡的主要原因。动脉粥样硬化由血管内皮功能障碍、脂质沉积、巨噬细胞吞噬、泡沫细胞形成、异常迁移以及血管平滑肌细胞(VSMCs)和基质细胞在促炎因子作用下增殖引起。在病理刺激下,如低密度脂蛋白(LDL)和甘油三酯水平、吸烟和肥胖,血管内皮细胞(ECs)的血管最内层被激活。单核细胞作为促炎细胞,可与活化的内皮细胞表达的黏附分子结合,通过趋化因子招募到内膜中。进入内膜后,单核细胞成熟为表达清道夫受体(scavenger receptor)的巨噬细胞,然后与脂蛋白结合并分化为泡沫细胞。在动脉粥样硬化病变过程中,血管平滑肌细胞(VSMC)最初通过产生细胞外基质蛋白(如胶原蛋白和蛋白多糖)来稳定斑块。几十年来,泡沫细胞积聚加上死亡细胞和濒死细胞碎片,导致动脉粥样硬化斑块进展。然而,在动脉粥样硬化晚期阶段,活化的巨噬细胞产生基质金属蛋白酶(matrix metalloproteinase)家族的酶,导致斑块破裂,从而引起心肌和脑梗死,这显著增加了动脉粥样硬化疾病的发病率和死亡率。最近的证据表明,VSMC也可以分化为巨噬细胞和间充质干细胞样细胞,促进病变扩大和不稳定。
表观遗传学由Conrad Waddington于1942年提出,是一种涉及遗传的表型,通过有丝分裂或减数分裂传递。从那时起,表观遗传学被多次重新定义。最近表观遗传性状被定义为“不改变DNA序列而由染色体改变引起的稳定可遗传表型”。表观遗传学的影响既有积极作用,也有消极作用。一方面,表观遗传调控因子可通过诱导转录抑制的肿瘤相关抗原de novo表达、增加新抗原表达和主要组织相容性复合体(MHC)加工/呈递、激活肿瘤免疫原性细胞死亡等机制协同促进肿瘤免疫原性;另一方面,如果暴露于化学物质、药物、应激或感染等环境因素,表观遗传学与免疫衰老导致的衰老细胞积累有关。
近年来,动脉粥样硬化除了是一种脂质沉积性和慢性炎症性疾病外,还被定义为一种表观遗传疾病。越来越多的证据表明,表观遗传修饰参与了动脉粥样硬化的发生和发展。基于微阵列的DNA甲基化分析显示,动脉粥样硬化患者的DNA甲基化水平高于健康对照。研究表明,表观遗传学不仅调控炎症细胞因子表达,还通过互作机制调控表观遗传修饰。然而动脉粥样硬化的慢性进展突出了动脉粥样硬化的异质性,到目前为止,斑块复杂环境中的特定细胞类型并不是动脉粥样硬化的唯一起始和驱动因子。相反,细胞类型的普遍效应受到表观遗传信号的严格调控和指导,而表观遗传信号又受到许多促动脉粥样硬化刺激的影响,包括LDL、促炎细胞因子和血液循环的物理力。此外,炎症分子通路(如Toll样受体(TLR)、NF-κB和JAK/STAT信号通路)与表观遗传修饰相关(图1)。因此,更好地理解表观遗传在动脉粥样硬化发病机制中的作用具有巨大的潜在转化价值。
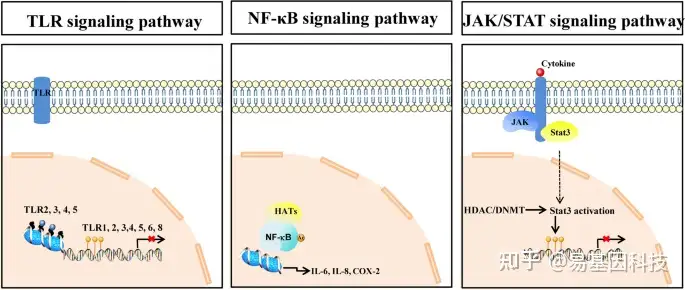
图1:表观遗传修饰和TLR、NF-κB、JAK/STAT信号通路相关。TLRs由DNA甲基化、组蛋白修饰等调控,最终导致TLRs表达变化。TLR基因启动子区(如TLR1、2、3、4、5、6、8等)的DNA甲基化可降低膜上TLR表达。根据修饰类型不同,TLR基因启动子附近核小体(Nucleosomes)区(如TLR2、3、4和5)的组蛋白修饰可正调控或负调控膜上TLRs表达;HAT直接与NF-κB互作或诱导其乙酰化,并将NF-κB招募到IL-6、IL-8和环氧合酶-2 (COX-2)等促炎基因的启动子区,调控炎症信号通路激活;JAK激酶应激后被激活,引起Stat3活化,导致Stat3核转移。Stat3也可由HDAC或DNMT激活,从而导致Stat3的DNA甲基化
本文就DNA甲基化和组蛋白翻译后修饰在动脉粥样硬化发生过程中的作用及其建立、维持和消除等方面的研究进展作一综述。还讨论了动脉粥样硬化过程中DNA甲基化和组蛋白翻译后修饰模式的新见解。
动脉粥样硬化中的DNA甲基化(DNA methylation in atherosclerosis)
DNA甲基化是最早发现的基因表观遗传修饰之一。与DNA甲基化相关的激活或抑制与甲基化环境和位点密切相关。DNA甲基化是一种动态可逆的修饰过程,是指在DNA甲基转移酶(DNMTs)作用下,胞嘧啶鸟嘌呤(CG)中的胞嘧啶转化为5-甲基胞嘧啶(5mC)的过程。在此过程中,S-腺苷甲硫氨酸提供一个甲基,而DNA去甲基化酶10 - 11易位蛋白(TET)将5mC转化为5-羟甲基胞嘧啶(5-hmC)。DNA甲基化分为三个阶段:建立(de novo DNA甲基化)、维持和去甲基化。在哺乳动物胚胎发育的早期阶段,DNA甲基化在DNMT3A/3B的作用下,主要发生在对称的5' -Cphosphate-G-3' (CpG)位点。两种主要的de novo DNA甲基化酶DNMT3A和DNMT3B在羧基末端包含一个高度保守的DNMT结构域(MTase domain)和两个染色质read结构域ATRX-DNMT3-DNMT3L (ADD)和PWWP。甲基化维持酶DNMT1与另一多结构域蛋白E3泛素蛋白连接酶UHRF1协同作用,UHRF1通过其SET- and RING-associated (SRA)结构域在复制叉头盒子(forkhead box, FOX)特异性结合半甲基化CpG二核苷酸。此外,TET甲基胞嘧啶双加氧酶(包括TET1、TET2和TET3)逐步氧化5mC为5-羟甲基胞嘧啶(5hmC)、5-甲酰胞嘧啶(5fC)和5-羧胞嘧啶(5caC),导致激活DNA去甲基化。
健康个体基因启动子区的CpG岛通常低甲基化,而非启动子区CpG岛则高甲基化。全基因组DNA低甲基化发生在非启动子区域,可导致在非准确区域转录启动和通常沉默位点的高转录活性。另一方面,整体DNA高甲基化通常会抑制表达、基因突变和等位基因丢失。在正常生理条件下,DNMTs和TETs协同维持DNA甲基化和去甲基化的平衡。研究表明,异常DNA甲基化与多种疾病相关,并在动脉粥样硬化中发挥重要作用,其中包括异常高甲基化和低甲基化。Zaina等使用全基因组重亚硫酸盐测序研究结果表明,与相应的健康对照组相比,主动脉粥样硬化区域的多个基因组位点均发生高甲基化。然而在动脉粥样硬化中存在全基因组低甲基化。既往研究表明,动脉粥样硬化斑块中以DNA低甲基化为主。在人类动脉粥样硬化晚期病变和ApoE敲除小鼠病变中,动脉粥样硬化发生基因组低甲基化。这些发现提示动脉粥样硬化中的DNA甲基化是一个动态过程,表现为早期升高,晚期降低。另一方面,DNA甲基化水平不仅与动脉粥样硬化的分期有关,而且与动脉粥样硬化的病变分级有关。通过全基因组DNA甲基化测序发现,在动脉粥样硬化的人主动脉中,DNA甲基化与动脉粥样硬化病变分级呈正相关。
DNA高甲基化和动脉粥样硬化(DNA hypermethylation and atherosclerosis)
越来越多的研究表明,DNA甲基化受炎症信号通路调控。例如用促炎刺激(如氧化低密度脂蛋白(oxLDL))处理人脐静脉内皮细胞(HUVECs)已被证明上调DNMT1并导致Krupel样因子2 (KLF2)基因启动子甲基化,从而导致KLF2受抑制和内皮炎症积聚。此外白细胞介素-6 (IL-6)可以鉴定DNMT1和DNMT3B蛋白稳定性,从而诱导基因的整体和启动子特异性DNA甲基化变化。同时,炎症信号通路受DNA甲基化调控,抑制DNMT3b可使转录因子叉头盒子P3(FOXP3)、转化生长因子-β、白细胞介素-10表达水平升高,白细胞介素-1β、干扰素- γ表达水平降低。在调控炎症因子过程中,DNA甲基化产生机制以及DNA甲基化在基因组三维结构中相关基因表达调控的机制尚未阐明。最近一项对542个人类转录因子研究发现,与未甲基化的转录因子相比,117个(22%)转录因子在甲基化时与其motif结合减少。通过阻止这些转录因子结合,DNA甲基化可以阻止包含其序列识别motif的CpG岛启动子的转录激活。
除炎症因子外,DNA甲基化还受其他刺激因素的影响。体外研究表明,暴露于紊流模式的内皮细胞具有更高水平的DNMT1,导致其基因组DNA高甲基化。肥胖与严重心血管或内分泌疾病(如动脉粥样硬化和卒中)的风险增加相关。在肥胖小鼠腹股沟白色脂肪组织中,Scara3表达降低。在2型糖尿病和动脉粥样硬化患者中观察到SCARA3高甲基化。此外,来自超重/肥胖的韩国受试者研究结果表明,TSPO相关蛋白1反义RNA 1 (TSPOAP1-AS1)启动子的DNA高甲基化与超重/肥胖相关,以及LDL胆固醇水平与TSPOAP1-AS1 DNA高甲基化水平显著正相关。这些启动子区域的高甲基化是否可能是动脉粥样硬化的潜在预测因素尚不清楚;因此需要进一步的研究。
动脉粥样硬化是一个涉及多种血管壁细胞和炎性细胞的复杂病理过程。内皮功能障碍是动脉粥样硬化的病理基础,并伴有血管壁通透性改变。导致脂质沉积、炎性细胞浸润以及平滑肌细胞迁移和增殖,进而发展为动脉粥样硬化。DNA甲基化异常引起的基因表达改变可导致细胞表型和功能改变。在动脉粥样硬化的关键因子氧化低密度脂蛋白(ox-LDL)处理的HUVECs中,DNMT3b介导的e1a刺激基因的细胞抑制因子(CREG)高甲基化,导致CREG表达抑制和内皮功能障碍。同型半胱氨酸(Homocysteine, Hcy)是动脉粥样硬化的独立危险因素,可上调10号染色体上的磷酸酶和张力蛋白同源物(phosphatase and tensin homologue on chromosome 10, PTEN)甲基化水平,并促进VSMC增殖,这是动脉粥样硬化发展的主要病理事件。线粒体融合蛋白2(mitofusin-2,MFN2)是线粒体外膜上一种重要的跨膜GTPase,其高甲基化可进一步促进hcy诱导的VSMC增殖。c-Myc与DNMT1启动子的结合增加是导致MFN2高甲基化的一种新的相关分子机制。Hcy也参与了动脉粥样硬化的炎症反应和DNA甲基化动态变化,通过促进SMAD7启动子高甲基化激活NF-κ b介导的血管炎症反应,且呈剂量和时间依赖性。DNMT1是体内外巨噬细胞炎症的决定因子,DNMT1通过抑制Kruppel样因子4 (KLF4)的表达促进巨噬细胞M1活化,并且DNMT1敲除的ApoE- /-小鼠可改善动脉粥样硬化形成并抑制斑块炎症。在一项对从紊流区分离的猪主动脉内皮研究中,研究人员发现,血流变化通过DNMT3a富集增加了KLF4启动子内CpG岛的DNA甲基化,从而导致动脉粥样硬化产生区域性影响。DNMT3b加速动脉粥样硬化,可能与人外周血调控性T细胞叉头盒子P3(FOXP3)高甲基化状态有关。在ApoE-/-小鼠中,Dnmt3b沉默通过减少病变大小和巨噬细胞含量,同时增加胶原和平滑肌细胞含量以减轻动脉粥样硬化。在人动脉粥样硬化斑块和动脉粥样硬化患者中,SMAD7启动子高甲基化,且与同型半胱氨酸水平和颈动脉斑块评分呈正相关。在非裔美国人样本中,针对心血管疾病危险因素进行校正后,AHRR、GFI1和LRRC52的DNA甲基化与动脉粥样硬化相关。这些发现表明,甲基化的SMAD7、AHRR、GFI1和LRRC52可能是动脉粥样硬化的新型预测生物标志物。
DNA低甲基化和动脉粥样硬化(DNA hypomethylation and atherosclerosis)
在细胞模型、动物和人类的许多研究中都揭示了DNA低甲基化的动脉粥样硬化。同型半胱氨酸的前体S-腺苷同型半胱氨酸(s-adenosylhomocysteine, SAH)诱导内皮细胞低甲基化,导致内皮细胞迁移增强,水通道蛋白1水平降低,水通透性受损,从而导致动脉粥样硬化发生。通过下调SAH水解酶水平还可通过降低DNMT1表达和p66Shc启动子甲基化激活调控氧化应激的关键蛋白p66Shc表达。然后诱导氧化应激损伤内皮功能,促进ApoE-/-小鼠动脉粥样硬化发生,提示SAH相关的内皮损伤可能促进动脉粥样硬化发生。抑制ApoE−/−小鼠SAH水解酶,通过抑制内皮细胞DNA甲基化,表观遗传上调Drp1表达,导致血管衰老和动脉粥样硬化。Dynamin相关蛋白1 (Dynamin-related protein 1, mdivi-1)作为一种Drp1特异性抑制剂,通过抑制线粒体mito-ROS/ NLRP3介导的M1极化来减缓ApoE-/-小鼠动脉粥样硬化。核糖核酸酶6在动脉粥样硬化患者外周血和斑块组织中表达上调。体内外研究表明,核糖核酸酶6启动子低甲基化加重了小鼠动脉粥样硬化,促进经oxLDL处理的小鼠主动脉VSMC增殖和迁移,并上调细胞中ROS含量和炎症因子分泌水平。此外与对照组相比,冠心病患者的IL-6启动子区域存在低甲基化,且在动脉粥样硬化患者中,IL-6基因的DNA低甲基化与IL-6基因表达增加相关。在动脉粥样硬化患者中,信号淋巴细胞活化分子7 (SLAM7)在晚期斑块中的表达显著高于早期动脉粥样硬化组织,在不稳定斑块中的表达显著高于稳定斑块。SLAM7高表达促进促炎细胞因子分泌并抑制VSMC增殖,是动脉粥样硬化的关键调控因子,可能是其潜在的治疗干预靶点。全基因组分析发现,与炎症时表达增加的非易损性病变相比,脂蛋白相关磷脂酶A2启动子区CpG位点(cg11874627)在动脉粥样硬化易损病变中的甲基化水平较低。总之,这些结果表明,异常的DNA去甲基化修饰(p66Shc、Drp1、核糖核酸酶6、IL-6、SLAM7和脂蛋白相关磷脂酶A2)参与动脉粥样硬化进展及其作为治疗靶点在动脉粥样硬化中的潜在作用。
TET1s缺失会加重振荡剪切流(oscillatory shear flow)诱导的动脉粥样硬化。DNA去甲基化酶TET2抑制促炎细胞因子、趋化因子的上调和炎症小体的激活,从而预防动脉粥样硬化。造血或髓系细胞特异性TET2缺失也会加重心力衰竭时的心功能障碍,这与NLRP3 /IL-1β通路激活相关。最近研究表明TET2通过调控beclin1依赖的自噬过程来改善ApoE-/-小鼠的动脉粥样硬化进展。
动脉粥样硬化中的组蛋白修饰(Histone modification in Atherosclerosis)
核小体(Nucleosomes)是染色质的功能单位,由147个DNA碱基对和一个由四组核心组蛋白(H2A、H2B、H3和H4)组装的八聚体组成。组蛋白N端延伸到核小体之外,可以通过乙酰化、甲基化、磷酸化、泛素化、糖基化和ADP-核糖基化修饰基因表达。这一过程被统称为组蛋白翻译后修饰。组蛋白修饰失衡可导致心血管疾病发生发展,而组蛋白H3、H4残基甲基化和乙酰化缺失是动脉粥样硬化的标志。在这些修饰中,组蛋白乙酰化和组蛋白甲基化是在炎症和心血管疾病中研究最多的修饰。
组蛋白甲基化(Histone methylation)
与组蛋白乙酰化相比,组蛋白甲基化在维持和形成异染色质结构、基因组印记、DNA修复、X染色质失活以及转录调控等方面发挥着更稳定的表观遗传标记作用。组蛋白甲基化主要发生在组蛋白H3和H4的赖氨酸(k)或精氨酸(R)残基上。根据不同位点的甲基化程度,可分为单甲基化(me)、二甲基化(me2)和三甲基化(me3)。组蛋白甲基化通常与转录抑制相关。
组蛋白甲基化过程主要由两种组蛋白甲基转移酶(HMT)催化。其中,组蛋白赖氨酸甲基转移酶(HKMT)包含6中组蛋白赖氨酸甲基转移酶复合体(KMT1-6,表1),组蛋白精氨酸甲基转移酶(protein arginine methyltransferase,PRMT)包含PRMT1、3、5、6和CARM1。组蛋白去甲基化酶大致分为两个家族:LSD(赖氨酸特异性去甲基化酶,包含LSD1和LSD2)和JMJD (JmjC结构域包含家族,包含KDM2, 3,4,5,6)。一般情况下,组蛋白H3和H4不同位点的甲基化以及甲基化程度对基因的转录调控具有重要意义。其中,H3K9me3、H3K27me3和H4K20me2/3介导转录抑制,而H3K4me1/2/3、H3K9me1、H3K27me1、H3K36me1/2/3和H3k79me1/2/3介导转录激活。
 表1:组蛋白甲基转移酶和去甲基转移酶
表1:组蛋白甲基转移酶和去甲基转移酶
动脉粥样硬化发生发展与M1型促炎表型分泌的炎症因子显著相关。瞬时受体电位A1 (Transient receptor potential A1)作为一种钙离子通透性非选择性阳离子通道,在动脉粥样硬化中过表达。它可以改变巨噬细胞中H3K27三甲基化水平,调控巨噬细胞向炎症表型方向发展。在单核细胞中,炎症细胞表现出H3K9和H3K27甲基化降低。冠心病患者CD14单核细胞中单核细胞趋化蛋白1 (monocyte chemoattractant protein 1, MCP1)血浆浓度显著上调,MCP1启动子的H3K9三甲基化降低。MCP1影响单核细胞趋化,是与动脉粥样硬化发展密切相关的关键趋化因子。
在促炎M1中观察到组蛋白赖氨酸甲基转移酶SETDB2 (KMT1家族成员)上调,而在造血细胞中SETDB2缺失会促进血管炎症和加速动脉粥样硬化。组蛋白H3K27甲基转移酶Ezh2通过抑制细胞因子信号转导抑制因子Socs3,以增加巨噬细胞炎症反应。髓系特异性Ezh2缺失可降低巨噬细胞泡沫细胞炎症反应,减少一氧化氮、IL-6和IL-12产生,从而减轻小鼠动脉粥样硬化。在VSMC特异性端粒沉默干扰因子1样(Dot1l)条件敲除小鼠模型中,Dot1l及其特异性诱导的H3K79me2直接调控Nf-κB转录,导致CCL5和CXCL10表达增加。小鼠Dot1l缺失可降低动脉粥样硬化斑块稳定性,并通过调控脂质生物合成基因程序促进炎性斑块巨噬细胞活化。这些发现提示DOT1L是动脉粥样硬化的潜在治疗靶点。
另一方面,赖氨酸去甲基化酶KDM4A/JMJD2A直接靶向oxldl诱导的巨噬细胞M1极化,而不依赖于NF-κB和HIF激活,这两个信号对巨噬细胞的促炎激活至关重要。LPS处理可促进HUVECs中JMJD3表达,并促进JMJD3核聚积。JMJD3减少了靶基因启动子区的甲基化状态,最终导致靶基因表达。
组蛋白乙酰化和去乙酰化(Histone acetylation and deacetylation)
组蛋白乙酰基变化是动脉粥样硬化的表观遗传学标记。乙酰化通过赖氨酸乙酰转移酶(KAT)将乙酰基从乙酰辅酶A转移到赖氨酸的ε-氨基侧链,这一过程可被KDAC逆转。在组蛋白结构中添加乙酰基会降低其正电荷和对负电荷DNA的亲和力,从而增加染色质的转录可及性。组蛋白乙酰化是研究最广泛的组蛋白修饰形式。组蛋白乙酰转移酶(Histone acetyltransferases, HAT)和组蛋白去乙酰化酶(Histone deacetylases, HDACs)在20世纪90年代中期至90年代末被发现。HATs和HDACs分别被重命名为KATs和赖氨酸去乙酰化基酶(KDACs),以区别于非组蛋白乙酰化。这两种酶通过组蛋白的可逆修饰来调控组蛋白乙酰化水平和基因转录。KATs分为3类:GCN5 (GCN5相关N -乙酰转移酶家族;包含GCN5和PCAF)、MYST (MOZ, MORF, Ybf2/Sas3, Sas2和Tip60)和P300 (CBP和P300)。KDAC分为4类:第1类(组蛋白去乙酰化酶(HDAC) 1、2、3、8),第2类(HDAC 4、5、6、7、9、10),第3类(SIRT 1 - 7)和第4类(HDAC11)。1、2和4类KDAC是典型的锌依赖性去乙酰化基酶,而4类KDAC是NAD依赖性sirtuin去乙酰化基酶。在人巨噬细胞中,LPS诱导p300募集并增强NADPH氧化酶5基因近端启动子内活性转录位点的组蛋白乙酰化,这表明对调控NADPH氧化酶5表达的表观遗传学通路的药理学靶向可能是动脉粥样硬化中一种值得关注的新治疗策略。
Ⅰ类KDAC(Class I KDACs)
越来越多证据表明HDACs参与了VSMC增殖和迁移的表型转换。最近的研究发现,HDAC1作为一种重要的调控因子,对主动脉VSMC迁移和表型转换至关重要。CD14 +单核细胞的调控因子X1缺失促进了MCP1启动子区域的H3和H4乙酰化和H3K9三甲基化,并通过减少HDAC1和杂化抑制因子3-9同源物1 (SUV39H1)的招募导致MCP1过表达。在内皮细胞中,组蛋白去乙酰化酶2 (HDAC2)保护内皮功能障碍和动脉粥样硬化。内皮-间充质转化(Endothelial-mesenchymal transition,EndMT)是动脉粥样硬化斑块不稳定的重要因素。动脉粥样硬化中HDACs与血管内皮稳定性及EndMT密切相关。HDAC3作为一种必需的促生存分子,对内皮祖细胞的分化至关重要。当HDAC3被敲低时,ApoE-/-小鼠发生动脉粥样硬化,血管破裂。HDAC3也影响动脉粥样硬化中的EndMT,在ApoE−/−小鼠和HUVECs中,HDAC3抑制剂通过调控炎症来抑制EndMT。在ox-LDL处理的HUVECs和ApoE−/−小鼠中,HDAC3通过microRNA-19b/PPARγ/NF-κB轴抑制炎症来保护动脉粥样硬化。
Ⅱ类KDAC (Class II KDACs)
HDAC4是一种关键的调控因子,参与多种细胞的增殖和迁移。HDAC4可促进VSMC增殖和迁移,干扰HDAC4可抑制VSMC增殖和迁移。此外,HDAC4还参与血管钙化(vascular calcification, VC)。最近,Abend等发现HDAC4在VC早期表达上调,并参与VSMC血管钙化和炎症反应。VC是一种发生于血管平滑肌细胞的活跃过程,以动脉内钙沉积为特征。它也与动脉粥样硬化的发病率和死亡率相关。而HDAC5在VSMC中作为促炎分子,由Nox4依赖的ROS产生和磷脂酰肌醇3-激酶(PI3K)/AKT通路介导。另一方面,HDAC6在动脉粥样硬化刺激下通过翻译后修饰上调,是CSEγ在血管内皮中表达的关键调控因子。在oxLDL处理的内皮细胞中,CSEγ表达减少,H2S生成减少,导致内皮功能障碍。胱硫醚γ-裂解酶通过抑制HDAC6活性保护血管内皮。在动脉粥样硬化中,靶向HDAC6可通过NF-κB/NLRP3通路减轻尼古丁诱导的巨噬细胞焦亡。
有动脉粥样硬化倾向的小鼠中表现出EndMT减少,斑块面积显著减少,而内皮特异性HDAC9基因敲除,而内皮特异性HDAC9调控EndMT和动脉粥样硬化斑块表型。Malhotra等发现HDAC9与腹主动脉钙化相关,并影响VSMC表型。在人主动脉VSMC中,HDAC9过表达促进钙化并降低收缩力,而HDAC9表达降低抑制钙化和增强细胞收缩力。HDAC9促进内皮-间质转化和不利的动脉粥样硬化斑块表型。HDAC抑制剂可抑制vsmc钙化,如apicidin、trichostatin、vorinostat和tubacin。
Ⅲ类KDAC (Class III KDACs)
SIRT6 (Sirtuin 6)是一种细胞核去乙酰化酶,在调控VSMC衰老和动脉粥样硬化中起关键作用。在人和小鼠斑块VSMCs中,SIRT6蛋白表达降低并受CHIP调控。SIRT6通过调控端粒维持和VSMC寿命进而抑制动脉粥样硬化发生,这一作用依赖于其去乙酰化酶活性。内源性SIRT6去乙酰化酶是VSMC衰老和动脉粥样硬化的重要抑制剂。
靶向DNA甲基化和组蛋白修饰治疗动脉粥样硬化
广泛的表观遗传修饰参与了动脉粥样硬化斑块的发生和发展。临床上,阿司匹林吸收会导致颅内动脉狭窄患者ABCB1甲基化水平降低。叶酸(缺乏叶酸会增加同型半胱氨酸水平)是一种动脉粥样硬化药物,在高脂饮食喂养的ApoE基因敲除小鼠中,叶酸可诱导内皮功能障碍,加速动脉粥样硬化病理过程,并增加血管过氧化物酶1、MCP1和血管内皮生长因子的DNA甲基化。因此,包括DNA甲基化和组蛋白翻译后修饰在内的表观遗传修饰是治疗包括动脉粥样硬化在内等多种疾病的一种有效方法。
以DNA甲基化为靶点的治疗策略
DNMT抑制剂是首批用于癌症治疗的表观遗传药物之一。靶向DNMT的药物包括胞嘧啶类似物、寡核苷酸类药物、DNA结合剂和S-腺苷甲硫氨酸抗性药物。胞嘧啶类似物可在DNA合成过程中不可逆地并入DNA中。当DNMT试图催化DNA甲基化时,这些胞嘧啶类似物DNMT可与DNMT共价结合,使DNMT不能与染色质分离,从而抑制活性。目前,美国FDA批准了两种胞嘧啶类似物DNMT,即5-氮杂胞苷(5-Aza-C)和5-Aza-CdR(商品名:地西他滨)。5-Aza-C用于治疗骨髓增生异常综合征,其不仅干扰DNA甲基化,还干扰mRNA合成,具有较强的毒性。通过5-Aza-C药理上调PTEN可减少斑块面积并保持体内SMC收缩蛋白表达。5-Aza-CdR适用于骨髓增生异常综合征和急性髓系白血病,它仅干扰脱氧核糖核酸(deoxyribonucleic acid)而不干扰核酸。5-氮杂-2'-脱氧胞苷抑制DNA甲基化可通过抑制巨噬细胞炎症来改善动脉粥样硬化。越来越多研究发现,在几种已建立的动脉粥样硬化动物模型中,5-Aza-CdR有效抑制了动脉粥样硬化进展,包括饮食诱导的ApoE−/−小鼠、LDLr−/−小鼠和接受颈动脉部分结扎的ApoE−/−小鼠动脉粥样硬化。
然而,对整体DNA甲基化的药理编辑缺乏特异性,可能导致不良反应,如自身免疫性疾病。在人动脉粥样硬化中,DNMT3B与CREG表达水平呈负相关,揭示了阻断CREG甲基化可能是治疗ox-LDL诱导的动脉粥样硬化的一种新方法。最近在RESCUE试验中,一种针对IL-6配体的全人源单克隆抗体Ziltivekimab被证明可以显著降低与动脉粥样硬化相关的炎症和血栓形成的生物标志物。此外,Ziltivekimab介导的IL-6配体抑制与较低的中性粒细胞-淋巴细胞比值相关,这一比值独立预测动脉粥样硬化事件,并且是残余炎症风险的潜在生物标志物,提示它可能破坏多种致动脉粥样硬化的炎症通路。另一方面,抑制Drp1降低了糖尿病ApoE-/-小鼠主动脉根部的巨噬细胞负荷、氧化应激和晚期钙化动脉粥样硬化斑块,以及人巨噬细胞产生的炎症细胞因子。Mdivi-1作为一种Drp1特异性抑制剂,是一种新型小分子PCSK9(proprotein convertase subtilisin/kexin type 9)抑制剂,是心血管预防的基石。
MG98等寡核苷酸类药物靶向DNMT的活性催化,阻止DNMT结合到特定基因的启动子以抑制其DNA甲基化。DNA结合剂(如SGI-1027)靶向DNMTs的辅因子结合位点。虽然尚未研究寡核苷酸药物、DNA结合剂和疾病之间的关系,但它们可能成为一种替代方法,因为它们不被纳入DNA,细胞毒性低。此外,一些中草药对动脉粥样硬化的DNA甲基化具有潜在的调控作用。一些草药和草药化合物,如姜黄素、栀子苷和白藜芦醇,也显示出在调控血管细胞中的表观遗传酶和动脉粥样硬化方面的前景。尽管目前的文献已经证明,一些天然存在的非核苷类DNMTi能够抑制小鼠动脉粥样硬化,但它们的具体作用机制和DNMTs的抑制作用在多大程度上参与动脉粥样硬化保护作用尚不清楚。目前仅少数用于治疗神经精神疾病的药物对组蛋白修饰酶或DNMTs有直接作用。重要的是,靶向胞苷类似物和非插入型甲基转移酶抑制剂的整体DNA甲基化已被FDA批准用于某些癌症的治疗。DNA甲基化是动态可逆的,具有个体差异和时空特异性。因此,针对特定个体和疾病特定阶段的DNA甲基化标志物设计特异性药物成为未来药物开发的挑战。
因此,靶向DNA甲基化通路可能是治疗动脉粥样硬化的有效方法,类似于目前DNA去甲基化药物在白血病中的临床应用。然而,我们需要进一步研究来破译细胞和基因特异性DNA甲基化变化(尤其是在人类中),并确定DNMT抑制剂治疗是否在疾病的不同阶段产生不同效果。
以组蛋白修饰为靶点的治疗策略
组蛋白甲基化通常与转录抑制相关。与其他表观遗传抑制剂相比,组蛋白甲基化抑制剂(HTMi)尚未得到广泛研究和开发。GSK126是一种有效的组蛋白甲基化抑制剂,对组蛋白N-甲基转移酶EZH2具有高度选择性。此外,它还能抑制H3K27me3,在mRNA和蛋白水平上显著降低促炎基因表达。
关于组蛋白乙酰化的潜在调控剂,garcinol和anacardic acid是具有组蛋白乙酰转移酶抑制剂(HATi)活性的天然化合物。Garcinol是一种从藤黄果实提取的的聚异戊烯基二苯甲酮,用于研究组蛋白乙酰化在调控早期生长反应蛋白1 (early growth response protein 1, EGR1)基因中的作用。近年来,一种新的果酸类似物MG149被开发为有效的、选择性的组蛋白乙酰转移酶(HATs) MYST家族(Tip60, KAT5和MOZ)抑制剂。MG149可抑制参与多种促炎细胞因子表达的NF-κB通路,在动脉粥样硬化等炎症性疾病中起关键作用。
组蛋白乙酰化与染色质开放和基因转录相关,HDACs可通过抗HATs影响组蛋白乙酰化。目前,组蛋白乙酰化的潜在调控因子HDAC抑制剂(HDACi)已被批准用于治疗血液系统恶性肿瘤,但其在动脉粥样硬化中的应用尚未在临床试验中进行研究。HDACi的药理作用是通过阻止靶基因启动子上的组蛋白去乙酰化来激活沉默基因。HDACi主要分为四类,包括环肽、脂肪酸和苯甲酰胺。Vorinostat (HDAC抑制剂)又称SAHA(suberoylanilide hydroxamic acid),是一种来源于异羟肟酸类的化合物。FDA已批准其用于T细胞皮肤性淋巴瘤的治疗,且作用于除Ⅲ类以外的所有HDACs。研究结果表明,SAHA以KLF2依赖性方式减少ApoE缺失小鼠的动脉粥样硬化病变大小。在Ldlr-/-小鼠中,另一种特异性HDACi曲古抑菌素A (TSA)通过增加清除率受体CD36启动子区、肿瘤坏死因子(TNF)- α和血管细胞黏附分子-1 (VCAM-1)的乙酰化和降低IL-6和il -1 β的表达来促进动脉粥样硬化。TSA可靶向C/EBPα/PPARγ轴,诱导C/EBPα乙酰化,从而缓解动脉粥样硬化。TSA在动脉粥样硬化中的不同作用可能是由于其非特异性,因为TSA对HDAC I, IIA和IIB具有抑制作用。这一现象提示在评估HDACi的药理作用时,必须研究HDAC抑制依赖和非依赖的机制。HDACi是一类很有前景的抗炎药物。近年来,丙戊酸(VPA)等I/IIa类选择性HDACi的高效给药系统被开发出来,以克服自由给药的常见缺点,如半衰期短和副作用。此外,它在原代人巨噬细胞中表现出抗炎作用,并能够减弱脂多糖诱导的炎症反应。TMP195是Ⅱa类HDAC的选择性抑制剂,在晚期动脉粥样硬化中,TMP195减少了关键的炎症通路,减轻动脉粥样硬化发生,从而为减少血管炎症提供了一种新的治疗策略。大多数现有或临床可用的HDAC抑制剂是通用的,选择性HDACi的开发可以减少其他靶点活性的副作用,如HDAC6的潜在通用毒性。罗米地辛(FK228)是一种针对HDAC1/2的选择性抑制剂,具有抗炎特性,并通过调控多种转录因子(krüppel-like因子5、CREB结合蛋白)的去乙酰化作用影响平滑肌细胞增殖。在ApoE−/−小鼠中,HDAC3特异性抑制剂RGFP966减轻了动脉粥样硬化病变,并抑制动脉粥样硬化斑块的EndMT。Bossche等使用特异性HDAC抑制剂研究结果表明,抑制HDAC3具有泛HDAC抑制剂的致动脉粥样硬化保护作用。由于部分减少了M1激活而没有增加泡沫细胞,在巨噬细胞中抑制HDAC,特别是HDAC3,显示出抗动脉粥样硬化的作用。在高脂饮食喂养的ApoE−/−小鼠中,Romidepsin通过增强STAT3乙酰化表观遗传调控VCAM-1表达,从而抑制动脉粥样硬化。由于DNA甲基化与组蛋白去乙酰化同时发生,因此联合应用DNA甲基化抑制剂和组蛋白去乙酰化抑制剂治疗炎症性疾病已成为研究热点。考虑到调控表观遗传沉默的现有化疗和正在开发中的其他药物可能会增加心肌梗死风险,因此靶向特定细胞的疗法可能是动脉粥样硬化治疗的替代方案。人类遗传学研究发现EndMT的主要调控因子ZEB2是冠状动脉疾病相关基因,ZEB2通过表观遗传抑制动脉粥样硬化中TGFβ和NOTCH信号通路来调控SMC表型转化。
用于动脉粥样硬化直接表观遗传治疗的纳米材料
以DNA甲基化或组蛋白修饰为靶点可能是动脉粥样硬化治疗的一个有效方法。然而如文中所述,表观遗传药物的许多靶点广泛表达,且一些表观遗传药物的生物利用度差、稳定性低、半衰期短,因此表观遗传治疗面临的主要挑战在于可能出现的副作用。纳米材料包括有机纳米颗粒(如聚合物纳米颗粒、脂质体、胶束和高密度脂蛋白纳米颗粒)和无机纳米颗粒(如金纳米颗粒、Fe3O4、介孔二氧化硅纳米颗粒和CuS),已被证明对动脉粥样硬化的治疗和诊断有效。近年来已经开发出大量具有物理和化学特性的纳米颗粒,它们能够有效地将表观遗传药物递送到患病细胞并调控其释放。例如由明胶酶、聚乙二醇(PEG)和聚-ε-己内酯组成的纳米颗粒特异性递送DAC,在小鼠胃癌异种移植模型中显著抑制肿瘤生长。用组蛋白去乙酰化酶抑制剂(iHDACs)功能化的聚合物纳米颗粒在间皮瘤肿瘤中产生最佳iHDACs释放,介导肿瘤重量显著且无毒性。另一方面,脂质体已被用作表观遗传药物的纳米载体:PEG功能化脂质体更有效地转运和释放一些抗肿瘤药物,包括HDAC抑制剂SAHA、PXD101和TSA。
结论
利用人类动脉粥样硬化组织或动物动脉粥样硬化模型来确定表观遗传学在血管发病机制中的作用,由于疾病的动态性质和组织异质性而变得复杂。表观遗传改变对多种细胞类型的普遍影响限制了其在疾病特异性或治疗中的临床应用。因此,系统地检测不同时期的表观遗传变化,能够制定全面的治疗策略。另一个面临的问题是,当多个翻译后修饰针对共有氨基酸残基(如赖氨酸残基)时,不同修饰之间可能存在竞争性拮抗。动脉粥样硬化相关细胞如何求同存异需要进一步的研究来适当地将表观遗传修饰因子加入动脉粥样硬化的治疗算法中。

参考文献:
Zhang L, Xia C, Yang Y, Sun F, Zhang Y, Wang H, Liu R, Yuan M. DNA methylation and histone post-translational modifications in atherosclerosis and a novel perspective for epigenetic therapy. Cell Commun Signal. 2023 Nov 29;21(1):344. pii: 10.1186/s12964-023-01298-8. doi: 10.1186/s12964-023-01298-8. PubMed PMID: 38031118.
相关阅读:
深度综述:人早期胚胎发育的表观遗传调控(染色质重塑+组蛋白修饰+DNA甲基化)
深度综述 | cfDNA甲基化诊断和监测肿瘤的研究进展与展望:胰腺癌
Cell|易基因微量DNA甲基化测序助力中国科学家成功构建胚胎干细胞嵌合体猴,登上《细胞》封面
Nature | 易基因DNA甲基化测序助力人多能干细胞向胚胎全能8细胞的人工诱导
标签:DNA,组蛋白,硬化,甲基化,动脉,粥样 From: https://www.cnblogs.com/E-GENE/p/17900755.html