大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
RNA甲基化是近年来研究基因表达调控转录后变化的重要研究领域,包括N6-甲基腺苷(m6A)在内的各种类型RNA甲基化参与人类疾病发展。MeRIP-seq作为一种新兴的在转录组范围内定量检测m6A水平的测序技术,拓展了RNA表观遗传学研究的基础和临床应用,且呈上升趋势。RNA甲基化数据分析的基本问题之一是通过对比病例和对照来鉴定差异甲基化区域(DMR)。现有开发了多种用于DMR检测的分析方法,但缺乏对这些分析方法的综合评估。
本文利用模拟数据和真实数据,全面评估了DMR calling的所有8种现有方法:FET-HMM、exomePeak(2.16.0版)、MeTDiff(1.1.0版)、DRME、QNB(1.0版)、exomePeak2(1.9.1版)、RADAR(0.2.4版)和TRESS(1.4.0版)。
模拟分析采用Gamma–Poisson模型和logit线性框架,并调试适应各种样本量和DMR比例进行基准检测。所有8种方法在低input水平区域中观察到低灵敏度,但样本量增加会大大提高灵敏度。TRESS和exomePeak2在检测精确度、FDR(False DiscoveryRate)、I型错误(type-I error)调控和运行时间等指标上表现最好,但受限于低灵敏度。DRME和exomePeak以FDR(假发现率)和I型错误(type-I error)膨胀为代价获得高灵敏度。三个真实数据集分析表明,这些方法在鉴定DMR长度和唯一发现区域有不同偏好性。
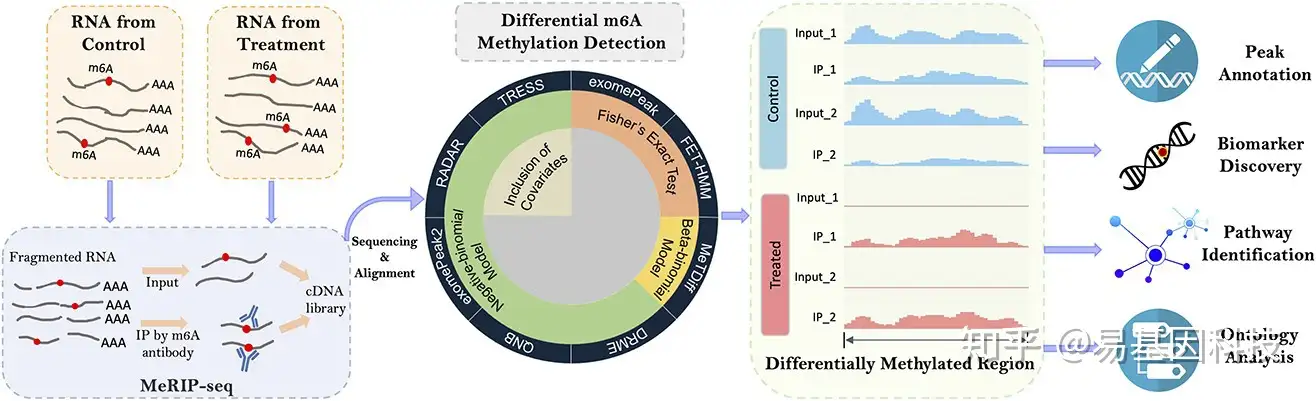
图1:MeRIP-seq实验和DMR检测示意图
MeRIP-seq从RNA样本生成配对的IP数据和input对照数据。将测序reads比对至参考基因组,然后通过最近开发的统计方法鉴定差异甲基化区域(DMR)。其核心统计模型和特征列于饼图内圆,下游对DMR基因进行peak注释、biomarker发现、通路(pathway)鉴定和基因本体(GO)分析。
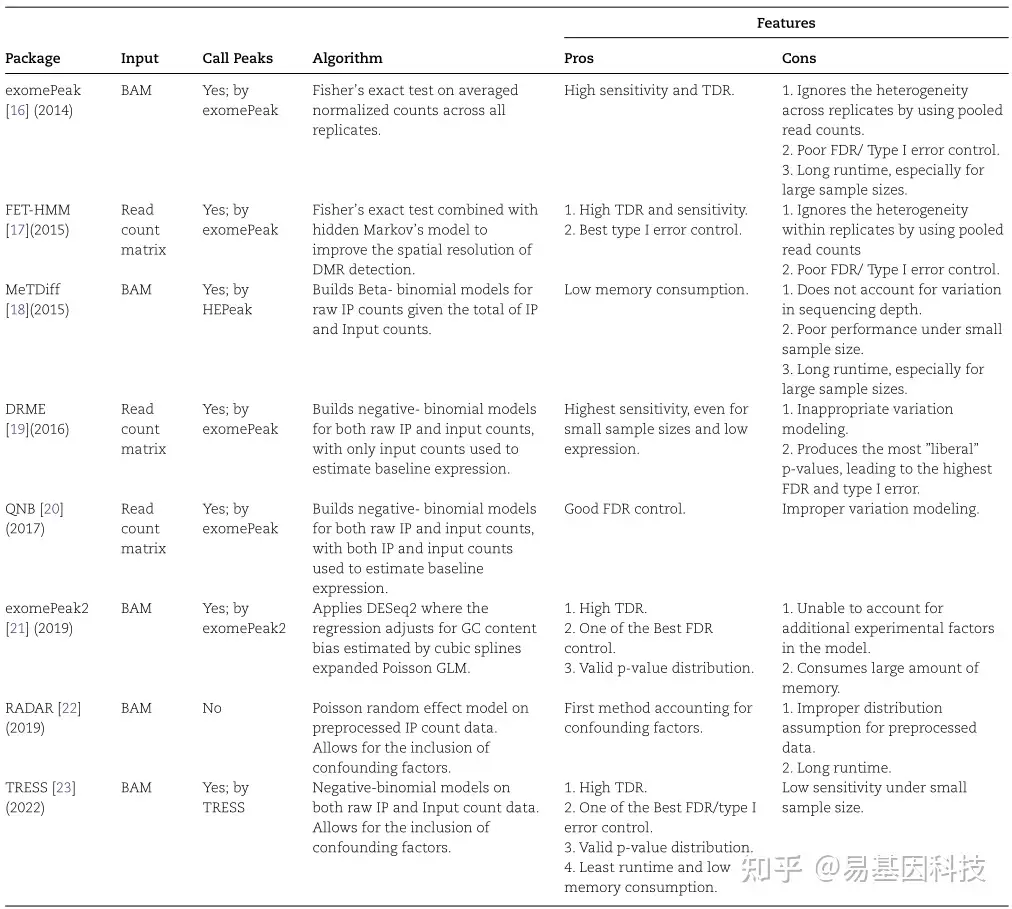
表1:现有差异RNA甲基化分析方法,方法按时间顺序排列。
TDR:真发现率(True Discovery Rate),即在某个截止点前被鉴定区域中所占的真阳性。
FDR:假发现率(False Discovery Rate)。
随着MeRIP-seq在过去几年的广泛使用,已经开发了几种检测DMR的计算方法,所有已知8种方法的详细信息见表1(按开发时间顺序排列)。作为第一个发布的工具,exomePeak在两种实验条件下对input对照和IP样本的归一化reads数应用Fisher精确检验(Fisher’s exact test,FET),因为使用所有重复的总reads,它忽略了生物学重复之间的异质性。随后改进exomePeak开发了MeTDiff和FET-HMM方法,MeTDiff假设β二项分布,并通过似然比检验(Likelihood Ratio Test,LRT)比较不同条件下的甲基化水平,然而MeTDiff没有很好解决测序深度的技术差异;FET-HMM采用了FET的改进版本,并使用FET的二值决策作为差异甲基化状态的观察,随后在检测到的甲基化区域内的小箱子上拟合隐马尔可夫模型(Hidden Markov Model,HMM),以合并沿基因组的依赖性,然而FET-HMM在合并每组中的重复检测,忽略了生物学重复之间的组内变化。2016年开发的DRME解决了这个问题,尤其是在小样本量的情况下,DRME假设IP和input对照计数数据均为负二项式模型,且仅使用input对照数据来预测背景基因表达,通过计算基于IP数据统计显著性来检测DMR。DRME的作者后来改进了他们的模型并开发了QNB,QNB也使用负二项式模型,与DRME不同的是,QNB在背景表达式的预测和检测统计的计算中结合了input对照和IP数据,DRME和QNB的共同限制是,两者都将ip内和input内的变化作为目标变化,但在MeRIP-seq中信号为IP/input比,应该严格建模该比方差。后来exomePeak的作者又提出了exomePeak2,与exomePeak相比,exomePeak2解释了IP效率和GC含量偏差变化,当存在多个重复时,exomePeak2 calling DESeq2通过将IP和input视为成对样本来鉴定DMR。
上述6种DMR分析方法仅适用于两组间的比较。在真实的生物学实验中,特别是在大型研究中,经常出现混杂协变量(如年龄或性别),但在上述方法中无法正确解释。为了解决这个问题,最近提出了两种方法:RADAR和TRESS,两种方法都使用线性框架将甲基化水平与实验因子相关联。RADAR采用泊松随机效应模型(Poisson random effect model),而TRESS采用伽马-泊松分布(Gamma–Poisson distribution)。TRESS与RADAR在两个方面不同,第一个区别是TRESS假设原始reads数遵循负二项式分布,通常用于建模各种测序数据类型。而RADAR假设预处理(从文库大小归一化开始,然后进行input对照调整)的计数数据遵循泊松分布,预处理后的数据不再是计数格式,因此泊松假设模棱两可。另一个区别在于一旦模型拟合,TRESS可以检测所有包含因子或其中任何线性组合的影响;而使用RADAR检测不同因子,需要重新提供设计矩阵并再次拟合模型,在计算上比较低效。总的来说,上面描述的方法列在表1中,显示了input数据类型、算法简要描述和使用中的各自优缺点。
数据生成模型和模拟(DATA GENERATIVE MODEL AND SIMULATION)
模拟框架的核心是伽玛-泊松分布,并为适应MeRIP-seq数据进行了适当修改。假设总共有10000个候选DMR,其中10%在处理和未处理的条件下差异甲基化。每种条件下的重复次数从2到10不等,以评估样本量对DMR calling的影响。
整体差异peaks比较(OVERALL DIFFERENTIAL PEAK COMPARISON)
对8种m6A DMR检测方法进行基准检测,每种场景下进行20次模拟。在一个共同实验设计下评估所有方法,作为整体比较基线,分别研究每个影响因子。
在每次模拟中,10000个候选DMR中的1000个被设计为真DMR,在病例组和对照组中都有三个重复。使用几个评估指标来评估八种DMR检测算法的性能,如使用真发现率(TDR)来分析生物标志物发现的精确度。还研究了ROC曲线(Receiver Operating Characteristic curve)、灵敏度和假发现率等经典指标。值得注意的是,FETHMM共有三种策略:“FHB”、“FHC”和“FastFHC”。本文使用FHC而非默认设置(FastFHC),因为默认设置中的编码有对P值的异常操作。
图2显示了在上述基线模拟场景中所有方法之间的DMR calling性能比较。如图2A所示,所有方法的TDR中,每个截止点,TRESS和exomePeak2的TDR值是最高的,且几乎相同。exomePeak、FETHMM、DRME和QNB生成的TDR相当,但略低于TRESS和exomePeak2。图2B显示TRESS、exomePeak2、exomePeak、FETHMM和DRME的AUC最高,而MeTDiff的AUC最低。值得注意的是,尽管TDR和ROC在方法比较中都是有效的指标,但TDR更具信息性,因为排名靠前的指标与生物标志物发现更相关。图2C中TRESS、exomePeak2、exomePeak、FETHMM和DRME的P值高度相关(Spearman相关性≥0.93),而MeTDiff得到的P值与其他方法差异较大。分析了最高和最低相关性的四对方法,并证明了FETHMM、exomePeak和exomePeak2之间的相似性。在图2D、E中,Benjamini–Hochberg调整后的P值计算灵敏度和假发现率,以0.05为截止值。尽管DRME、FETHMM和exomePeak具有高灵敏度,但其FDR值也很高。结合这两个指标,表明DRME、FETHMM和exomePeak的I型错误(type-I error)膨胀。MeTDiff和QNB在模拟中表现出不稳定性。MeTDiff很难鉴定出真阳性,因此并不是在所有的比较中都表现良好。TRESS和exomePeak2实现了几乎相同的最佳整体性能,在保持低FDR的同时发现了相当一部分真阳性。图2F中还总结了使用平均灵敏度和FDR的联合分布的总体性能。理想情况下,好的方法应该具有高灵敏度,同时保持低FDR,因此位于左上角区域的方法是首选方法。
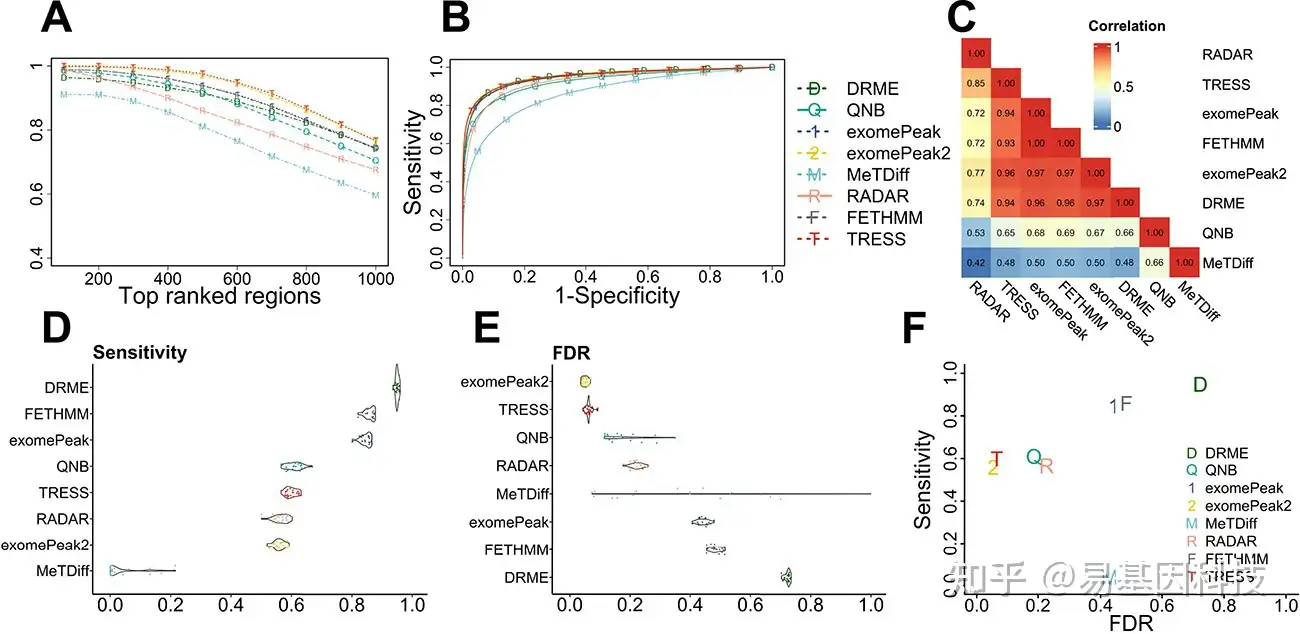
图2:m6A-DMR检测方法的性能比较。
- 每种方法鉴定的排名靠前区域的真发现率(TDR)。TDR定义为调整后p值排名前靠前区域中真DMR占比。
- DMR检测方法的受试者工作特征(ROC)曲线。
- 8种方法的p值相关性热图。
D-E. 每种方法的灵敏度和FDR分布的小提琴图,用BH调整后的p值计算。
F. 每种方法检测DMR的平均灵敏度与FDR。模拟在三个病例组、三个对照组、10%真DMR的情况下进行。N=20次模拟。
样本量(SAMPLE SIZE)
接下来研究样本量对DMR calling精确度的影响,因为样本量通常是实验设计中的主要参数。本研究两组的模拟样本量分别为2、3、5、7和10,每种条件下2、3、7、10个样品的TDR分别如图3A-D所示。几乎所有方法在靠前排名 (如前100或前200)calling区域获得高TDR(>0.8),且在排名靠后时显示出精确性下降。具体来说,TRESS和exomePeak2在所有截止点上都保持最高的精确度,而MeTDiff表现最差,沿秩递减的精确度最低。随着样本量增加,所有方法的精确度都有所提高。当N=7和10时,这种趋势尤其明显,其中方法报告的TDR值相似。在图3E中,TDR以热图的形式呈现,包括所有模拟场景下的结果(N=2、3、5、7、10),按排名前400、700、1000和1500区域进行分层。总体而言,所有方法中TDR值随区域排名提高和样本量增加而增加。大样本量可以大大提高检测精确度,即使是排名中等区域(如前1000名)。RADAR和MeTDiff在小样本量中 (N=2和3)的检测精确度较低,但随着样本量增加,其性能几乎相同。即使在极小的样本量下(N= 2), TRESS和exomePeak2的TDR也大于0.8。在经验贝叶斯框架(empirical Bayes framework)下,TRESS和exomePeak2在全基因组中实现了信息借用,因此其在小样本量中的表现优于其他方法。在其他基因组学研究中,这种建模技术已被证明是有效的统计框架,特别是对于小样本量。总之,对于小样本量的项目,TRESS和exomePeak2是首选。
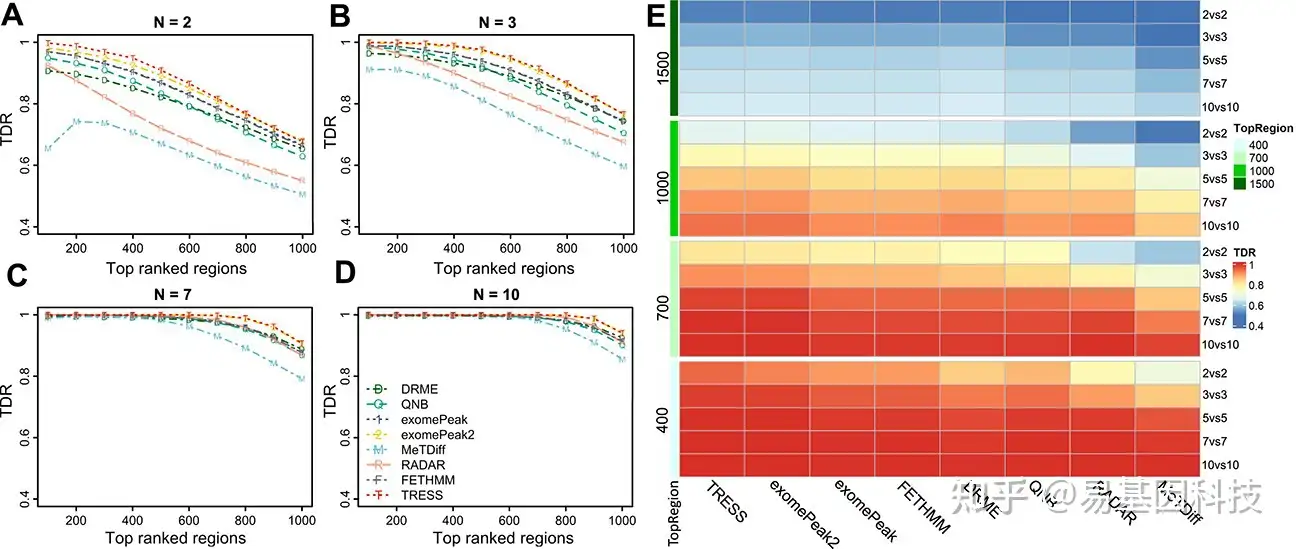
图3:不同样本量的DMR检测精确度比较。
A-D. 每组中进行2、3、7和10次重复的样本量下,每种方法鉴定的排名靠前区域的真发现率(TDR)。
E. 不同样本量和TOP区域截止值组合下的TDR值热图。样本量标注在右侧,每组2个、3个、5个、7个和10个。排名靠前区域截止线标注在左侧,范围从前400名、前700名、前1000名到前1500名。方法在热图中按列排序。在10%真DMR下进行了N=20次模拟,取平均TDR值。
分层评估(STRATIFIED ASSESSMENT)
高通量测序数据(如批量RNA-seq)的差异表达分析准确性高度依赖于表达水平,因此本研究按input范围分层检测DMR准确性。根据input对照分布,候选区域根据其平均input计数分为五层:第一层1(0~10)、第2层(10~20)、第3层(20~40)、第4层(40~80)和第5层(80~+∞). 以0.05值为标称值显著性水,所有方法在5个分层中的灵敏度和FDR如图4所示。方法按各层的平均值排序,当从较低分层转到较高分层时,所有方法都提高了灵敏度(图4A–C),低input区域通常容易受模拟噪声影响。即使在第一层,DRME也具有较高灵敏度(>0.75),且在所有区域中具有相对较好性能。随着样本量增加,DRME灵敏度仍在提高。随着样本量增加,所有方法都表现出增加和减少的可变灵敏度,且这种性能增益对于较低层非常显著,表明大样本量有助于更可靠预测,尤其是对于受高背景噪声影响更大的区域。其中,exomePeak2从样本量的增加中受益最大,从第七位上升到第四位。对于FDR的结果,更大样本量不如灵敏度(图4D–F)。TRESS和exomePeak2在所有分层和样本量大小中显示出较小且最一致的假发现率(FDR)。在小样本量下(N=3),MeTDiff在较低input区域的FDR较差,而随着样本量增加,FDR得到很大的提升。exomePeak、FETHMM和DRME受较差FDR影响,即使在大样本情况下也是如此(N=10)。
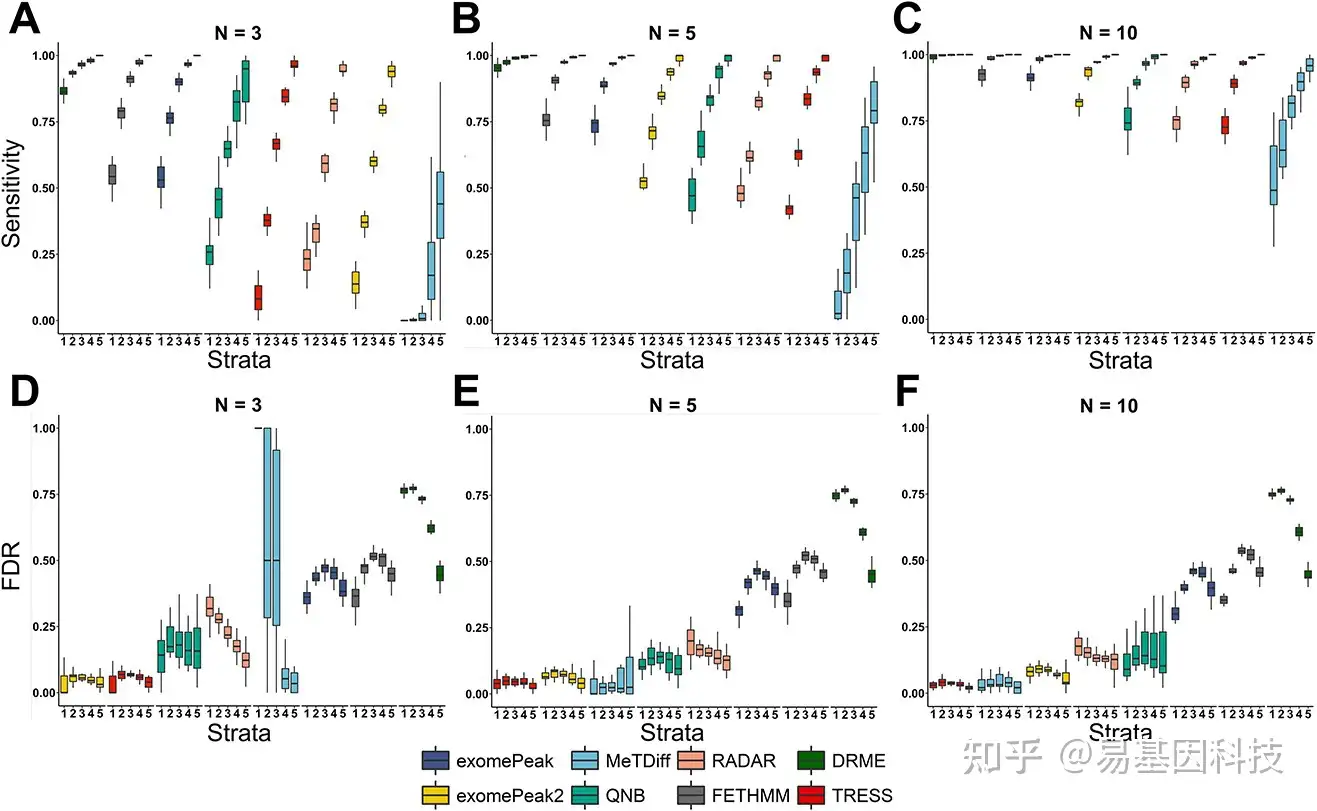
图4:按平均input计数值分层分析灵敏度和FDR。灵敏度和FDR以BH调整后P值计算,以0.05为截止值确定显著性。
A–C. 分层灵敏度,每组分别设置3个、5个和10个重复。
D–F. 分层FDR,每组分别设置3个、5个和10个重复。在10%DMR下进行N=20模拟。
I型错误和p值有效性(TYPE I ERROR AND VALIDITY OF P-VALUES)
为了研究8种方法的I型错误和p值有效性,在null条件下进行假设模拟,其中没有(0%)候选区域存在差异甲基化。使用bh调整后的p值以0.05标称值为显著性水平获得每种方法鉴定的DMR。在每组设置2、3、5、7和10个重复的情况下,计算经验I型错误率(表2)。在所有情况下,TRESS和FETHMM的I型错误率都接近0.05,表明其I型错误率接近标称值。exomePeak2更为“保守”,小样本下(N = 2、3)以低灵敏度获得最佳FDR(图2D、E)。DRME是最“自由”方法,与其高灵敏度和FDR相匹配(图2D、E)。
P值有效性分析检测了null条件下P值是否均匀分布在0和1之间,并在图5中说明了每组使用三个重复的结果。在QQ图(Quantile-quantile plot)中,TRESS和exomePeak2产生的p值与预期值最为一致(图5A,位于或接近对角线参考线)。大多数方法生成自由p值(右下方区域),而FETHMM在大多数区域过于保守(左上方区域)。由于小p值在DMR检测中更具信息性,本研究还应用-log10转换,重点关注图5B中小p值分布。TRESS、exomePeak2和RADAR表现最好,而其他方法提供的p值过小,表明I型错误膨胀。且对样本量不敏感(图2E),其中TRESS、exomePeak2和RADAR产生的的FDR控制得最好,也最稳定。
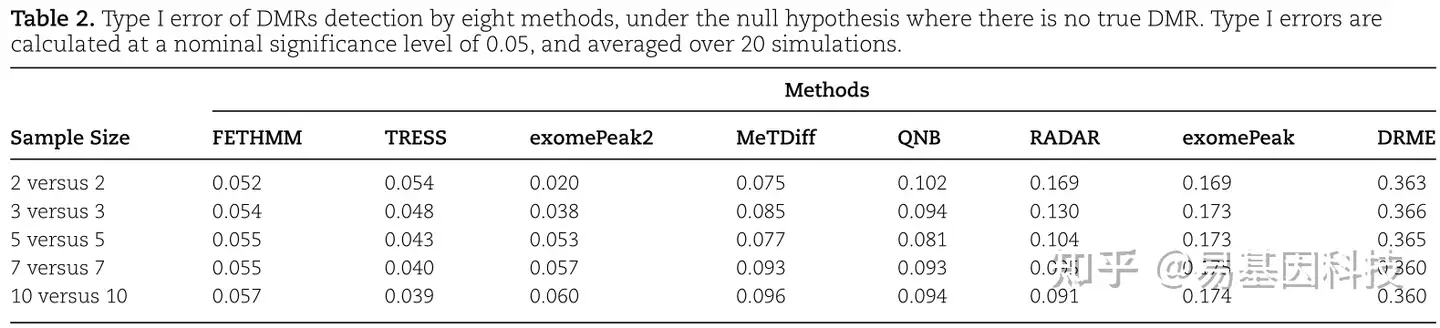
表2:在无真DMR的null假设下,八种方法检测DMR的I型错误(0.05标称值显著性水平计算,并在20次模拟中取平均值)
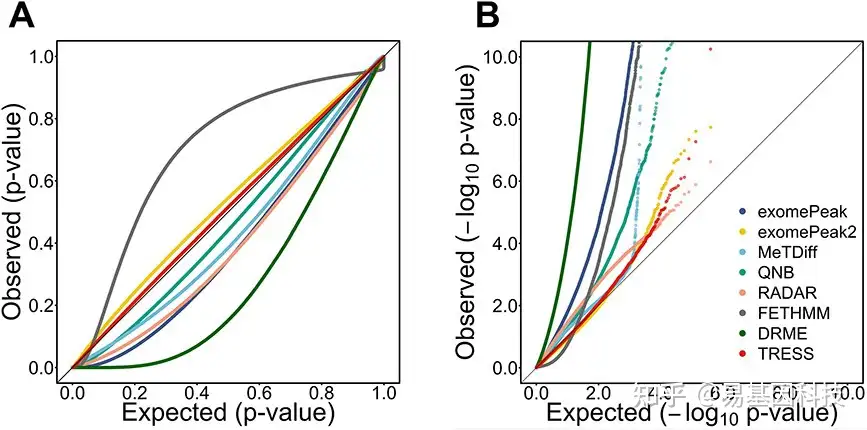
图5:分析null条件下模拟观察到的p值有效性。
- QQ图(Quantile–quantile plot)将p值分布与null下的期望分布U(0,1)进行比较。
- QQ图进行−log10转换,重点关注小P值。在无DMR的null假设下进行20次模拟。样本量N=3 /组。
运行时长和内存消耗(RUNTIME AND MEMORY CONSUMPTION)
BAM文件为默认input评估每种方法的软件运行时间和计算内存消耗。基于同一节点、同一内核和200 GB内存的高性能计算(HPC),在不同样本量下,五种方法的运行时长如图6A所示。随着样本量增加,所有方法都显示出更长的运行时间。与其他方法相比,TRESS和exomePeak2的运行时长都更短,且随着样本量增加更为明显。exomePeak和MeTDiff在所有样本量中具有相似的运行时间。RADAR的运行时间最慢。由于大多数方法都将BAM文件作为标准input,因此进一步对计算内存消耗进行了基准检测(图6B)。MeTDiff和exomePeak消耗内存最少(分别为3.81 GB和4.62GB)。TRESS消耗的内存略多于MeTDiff和exomePeak。exomePeak2利用了最多的内存(170.28GB)。模拟在HPC中进行,每个方法calling都有1个节点、40个内核和200 GB可用内存。
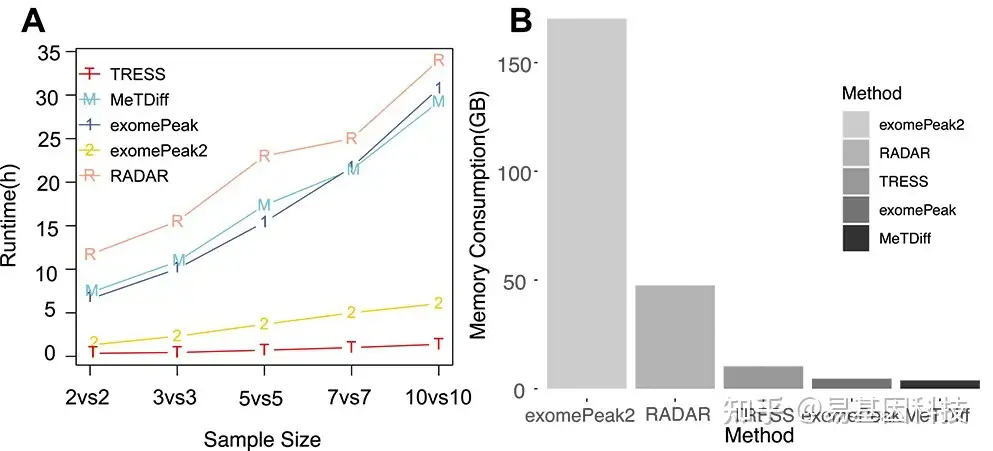
图6:m6A DMR检测方法的运行时长和内存消耗比较。
- 五种不同方法在不同样本量下的运行时长比较,以小时为单位。
- 计算五种不同方法的内存消耗,单位为GB。
真实数据分析(REAL DATA ANALYSIS)
首先从一项研究METTL3-METTL14复合体介导哺乳动物核RNA m6A甲基化的研究中获得了真实数据集(GSE46705),将其标记为“RD1”。在该研究中,人类HeLa细胞系有四种样品类型:一种野生型(WT)样品和三种处理过的样品,这些处理对应于复合体METTL3、METLL14和WTAP的敲除(KD)。每个样品2个重复。将TRESS、exomePeak、exomePeak2、MeTDiff和RADAR方法应用于该真实数据,以鉴定m6A差异甲基化。同时还采用了适用于分析MeRIP-seq数据的MACS3方法。MACS3已被先前的几项研究表明其作为MeRIP-seq数据差异分析的有效工具的潜力。该研究只分析以BAM文件为input比较的方法,因此排除了QNB和DRME等以reads计数矩阵为input的方法。
原始FASTQ文件被比对到人类参考基因组hg18,使用带有默认参数的STAR标准流程。比对后的BAM文件作为所有五种方法的input进行比较,主要分析WT和METTL3样品之间的差异甲基化。DMR calling在FDR<0.05的显著性水平上进行。在过滤掉短(宽度<150)和重叠区域后,TRESS、exomePeak、exomePeak2、MeTDiff和RADAR分别鉴定出1413、1397、5272、161和2924个DMR。exomePeak2鉴定出最多的DMR,而MeTDiff鉴定的DMR最少。
五种方法分析WT组与METTL3组真实数据的性能比较如图7所示。使用ChIPseeker对DMR进行注释(图7A)。结果显示,除了RADAR以外的大多数方法都支持3'UTR的DMR。RADAR偏好基因下游外显子区(即非第一外显子)。所有方法的启动子和下游外显子区均表现出相当数量的组成基因组表征。图7B显示了五种方法的5个重叠区域。exomePeak2发现3348个特异性DMR,是所有DMR中最高的。两种方法之间重叠区域的最高数量是由exomePeak和exomePeak2 calling的1038个重叠,而两种方法间重叠区域的最少数量是由TRESS和MeTDiff calling的15个重叠。DMR的peaks宽分布(log scale)如图7C所示。TRESS偏好150–400bp中长区域,RADAR具有双峰分布(bimodal distribution),覆盖中长和长两个区域。鉴定出1038个共有区域的FDR(图7D)。与exomePeak2相比,exomePeak是一种更保守的方法。同时由TRESS、exomePeak、exomePeak2、MeTDiff和RADAR方法显示了WT和METTL3样品之间共有DMR的两个示例(图7E),这两个区域覆盖蛋白编码基因TEX264(chr3)、PRICKLE4、TOMM6和USP49(chr6)。先前的研究表明,TEX264能够激活信号受体活性,并参与蛋白-DNA共价交联修复。USP46通过剪接体参与半胱氨酸型内肽酶活性、组蛋白H2B保守的C-末端赖氨酸去泛素化和mRNA剪接。对exomePeak2和RADAR进行GO(Gene Ontology)通路分析(图7F),在exomePeak2的DMR中,前三个GO富集是“生长因子受体和第二信使的信号通路疾病(Diseases of signal transduction by growth factor receptors and second messengers)”、“TP53转录调控(Transcriptional regulation by TP53)”和“I类MHC介导的抗原处理和呈递(Class I MHC mediated antigen processing & presentation)”。
同时在另外两个真实数据集(GSE94613和GSE115105)中进行检测,并将它们标记为“RD2”和“RD3”,其中, “RD2”包括12个METTL3敲低细胞系和对照的人类样本,“RD3”包括两个Ythdf1敲低和对照的野生型小鼠骨髓来源树突状细胞(BMDC)。对这两个数据集进行相同的分析,根据DMR数量和3’UTR在三个真实数据集中的百分比对五种方法进行排序(图7G)。exomePeak2软件calling了三个真实数据集中最多的DMR,其次是RADAR。在3’UTR方面,除了exomePeak2和MeTDiff之间的微小差异外,数据集之间再次观察到一致结果(图7H)。
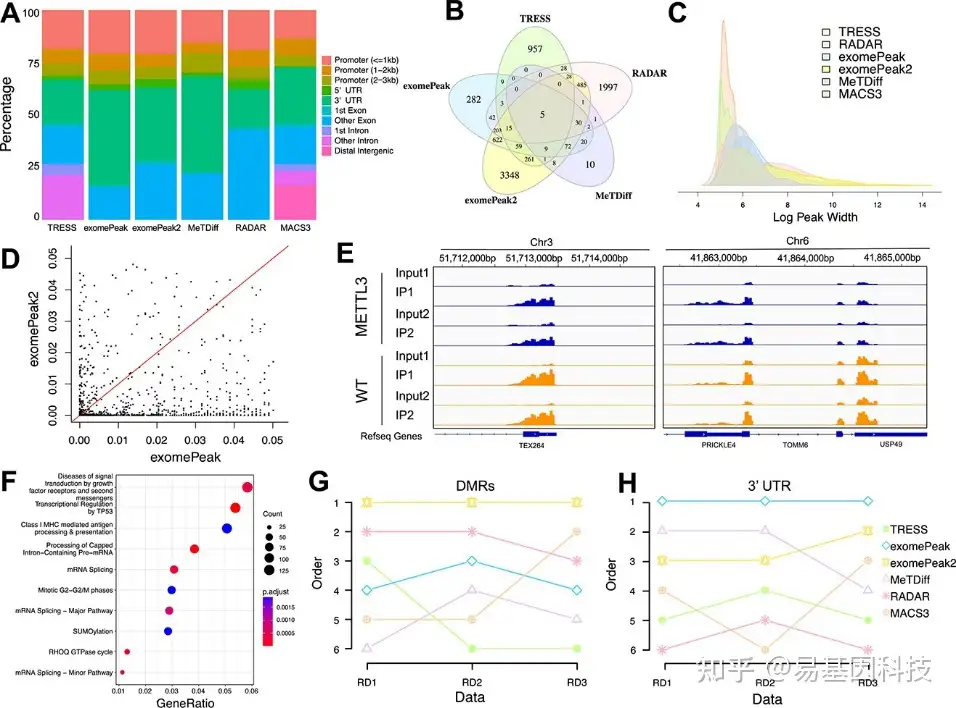
图7:真实数据的差异m6A甲基化方法。
- 条形图显示在已鉴定的DMR中各种基因组特征分布。TRESS、exomePeak、exomePeak2、MeTDiff、RADAR和MACS3采用相同的FDR 0.05截止值来calling显著性。
- 维恩图显示通过五种方法鉴定的DMR重叠。
- 六种方法的峰宽分布密度图(log scale)。
- exomePeak和exomePeak2的1038共有区域的成对FDR值散点图。
- 共有DMR的peaks差异分析可视化的两个例子。所有差异peaks分析均在野生型(WT)组和METTL3组之间。
- exomePeak2的DMR基因的GO分析,显示DMR数最多。
- 三个真实数据集中DMR计数排序。
- 三个真实数据集中3’UTR百分比排序。
比较要点(Key Points)
- 新型表观转录组测序技术能够使用数据驱动的方法评估RNA修饰。
- 差异表观转录组分析需要对成对的input对照和IP样本进行适当的建模,以适应技术和生物噪声、peaks值检测并解决小样本量问题。
- TRESS和exomePeak2在基准研究中表现出高TDR、低FDR和超高灵敏度。
- 检测精确度可能会受低input表达影响,但受益于样本量增加。
- RADAR、TRESS和exomePeak2显示了顶级严格的I型错误控制和null下的有效p值分布。MeTDiff计算内存消耗最少,TRESS运行时间最快。
关于易基因RNA m6A甲基化测序(MeRIP-seq)技术
易基因MeRIP-seq技术利用m6A特异性抗体富集发生m6A修饰的RNA片段(包括mRNA、lncRNA等rRNA去除所有RNA),结合高通量测序,可以对RNA上的m6A修饰进行定位与定量,总RNA起始量可降低至10μg,最低仅需1μg总RNA。广泛应用于组织发育、干细胞自我更新和分化、热休克或DNA损伤应答、癌症发生与发展、药物应答等研究领域;可应用于动物、植物、细胞及组织的m6A检测。
大样本量m6A-QTL性状关联分析,传统MeRIP单个样品价格高,通常难以承担。易基因开发建立MeRIP-seq2技术,显著提成IP平行性,实现不同样本间相对定量,降低检测成本。
易基因提供适用于不同科研需求的MeRIP技术:
- m6A甲基化-常量mRNA 甲基化测序(MeRIP-seq)
- m6A甲基化-常量mRNA +lncRNA甲基化测序(lnc-MeRIP-seq)
- m6A甲基化-微量mRNA +lncRNA甲基化测序(Micro-lnc-MeRIP-seq)
- 高通量m6A甲基化-常量mRNA甲基化测序(MeRIP-seq2)
技术优势:
- 起始量低:样本起始量可降低至10-20μg,最低仅需1μg总RNA;
- 转录组范围内:可以同时检测mRNA和lncRNA;
- 样本要求:可用于动物、植物、细胞及组织的m6A检测;
- 重复性高:IP富集重复性高,最大化降低抗体富集偏差;
- 应用范围广:广泛应用于组织发育、干细胞自我更新和分化、热休克或DNA损伤应答、癌症的发生与发展、药物应答等研究领域。
研究方向:
m6A甲基化目前主要运用在分子机制的理论性研究
- 疾病发生发展:肿瘤、代谢疾病(如肥胖/糖尿病)、神经和精神疾病(如阿尔兹海默症/抑郁症)、炎症…
- 发育和分化:早期胚胎发育、个体/组织/器官生长发育、干细胞分化与命运决定、衰老
- 环境暴露与响应:污染、抗逆、生活方式
关于m6A甲基化研究思路
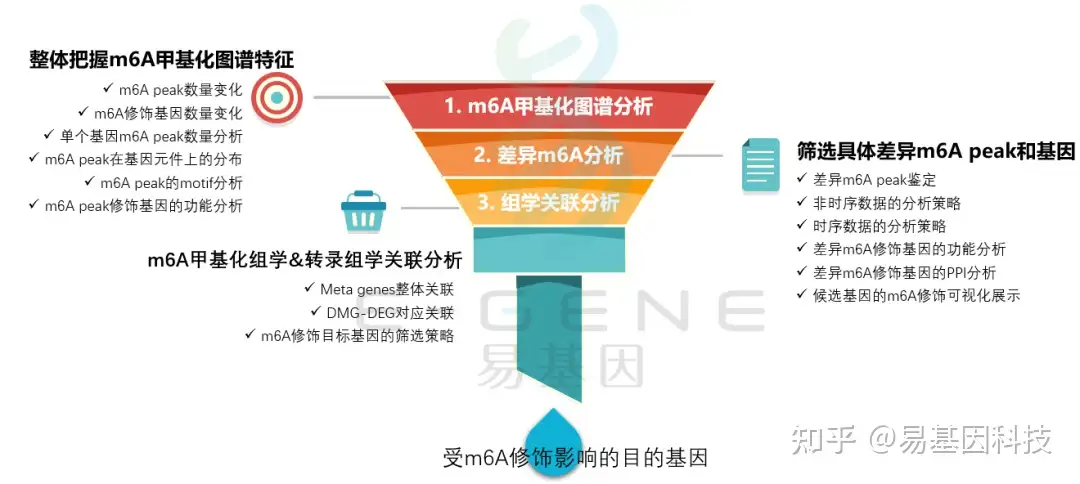
(1)整体把握m6A甲基化图谱特征:m6A peak数量变化、m6A修饰基因数量变化、单个基因m6A peak数量分析、m6A peak在基因元件上的分布、m6A peak的motif分析、m6A peak修饰基因的功能分析
(2)筛选具体差异m6A peak和基因:差异m6A peak鉴定、非时序数据的分析策略、时序数据的分析策略、差异m6A修饰基因的功能分析、差异m6A修饰基因的PPI分析、候选基因的m6A修饰可视化展示
(3)m6A甲基化组学&转录组学关联分析:Meta genes整体关联、DMG-DEG对应关联、m6A修饰目标基因的筛选策略
(4)进一步验证或后期试验
易基因科技提供全面的RNA甲基化研究整体解决方案。
参考文献:
Duan D, Tang W, Wang R, Guo Z, Feng H. Evaluation of epitranscriptome-wide N6-methyladenosine differential analysis methods. Brief Bioinform. 2023 May 19;24(3) pii: 7111718.
相关阅读:
14种全基因组DNA甲基化测序(WGBS)标准分析比对软件的比较 | 生信专区
干货分享 | DNA甲基化差异水平分析中的DMC、DMR、DMG鉴定
全基因组CpG密度和DNA甲基化分析方法比较(MeDIP、RRBS和WGBS)| 研究综述
标签:seq,甲基化,MeRIP,exomePeak2,TRESS,m6A,DMR,样本量 From: https://www.cnblogs.com/E-GENE/p/17570791.html