大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
超级增强子(Super enhancer)是一类包含多个普通增强子的大簇,主要富集高密度的转录因子、辅助因子及增强子相关表观修饰位点。与普通增强子相比,超级增强子具有更高的转录激活能力,在细胞类型特异性发育、分化以及肿瘤的发展进程中发挥关键作用。
超级增强子的鉴定是依据增强子转录活性标记分子结合水平强度的差异,这些分子包括辅因子 (如Mediator、cohesin)、组蛋白修饰标记 (如H3K27ac、H3K4me1)、染色质修饰分子 (如p300) 和BET家族蛋白(BRD4)等。当前超级增强子的鉴定主要通过染色质免疫沉淀测序(ChIP-seq),首先通过ChIP-Seq分析这些增强子转录活性标记分子在基因组上的富集情况,确定活性增强子位点。之后再对所有活性增强子进行分析,鉴定得到超级增强子。通过与转录组测序数据进行关联分析,可发现超级增强子所调控的靶基因。
2021年11月4日,武汉大学吴旻教授课题组在《Nat Commun》杂志发表了题为“Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers”的研究论文,该研究通过分析结直肠癌(CRC)病人的肿瘤和癌旁组织的增强子和转录组变化,鉴定出多种CRC相关增强子和转录因子,为结直肠癌的研究提供了重要的表观基因组资源和候选研究对象。

标题:Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers结直肠癌的全基因组分析鉴定出PHF19和TBC1D16是致癌的超级增强子
时间:2021.11.04
期刊:nature communications
影响因子:IF 17.694
技术平台:ChIP-seq、全基因组测序、RNA-seq等
研究摘要:
结直肠癌(CRC)是最常见的癌症之一。尽管CRC的基因组突变和单核苷酸多态性已被广泛研究,但结直肠癌患者组织中的表观基因组状态仍不了解。
本研究将基因组学与转录组学分析相结合,对73对结肠直肠癌配对患者组织(同一患者的肿瘤和癌旁组织)在全基因组水平上进行多组学测序,共产生147个H3K27ac的ChIP-Seq样本、144个RNA-Seq样本、147个全基因组测序样本和86个H3K4me3 ChIP-Seq样本。
分析鉴定出结直肠癌中5590个获得的差异增强子位点(gain VELs)和1100个缺失的差异增强子位点(lost VELs),334个获得的差异超级增强子位点(gain VSELs)和121个缺失的差异超级增强子位点(lost VSELs)。
通过motif分析和核心调控回路分析预测结直肠癌中的多个关键转录因子。进一步的实验验证了调控PHF19和TBC1D16的超级增强子在调控结直肠癌肿瘤发生中的作用,鉴定出KLF3是结直肠癌的致癌转录因子。本研究为结直肠癌的表观遗传学研究提供了重要的表观基因组资源和功能因子。
结果图形
(1)结直肠癌患者组织中增强子分布的全基因组研究
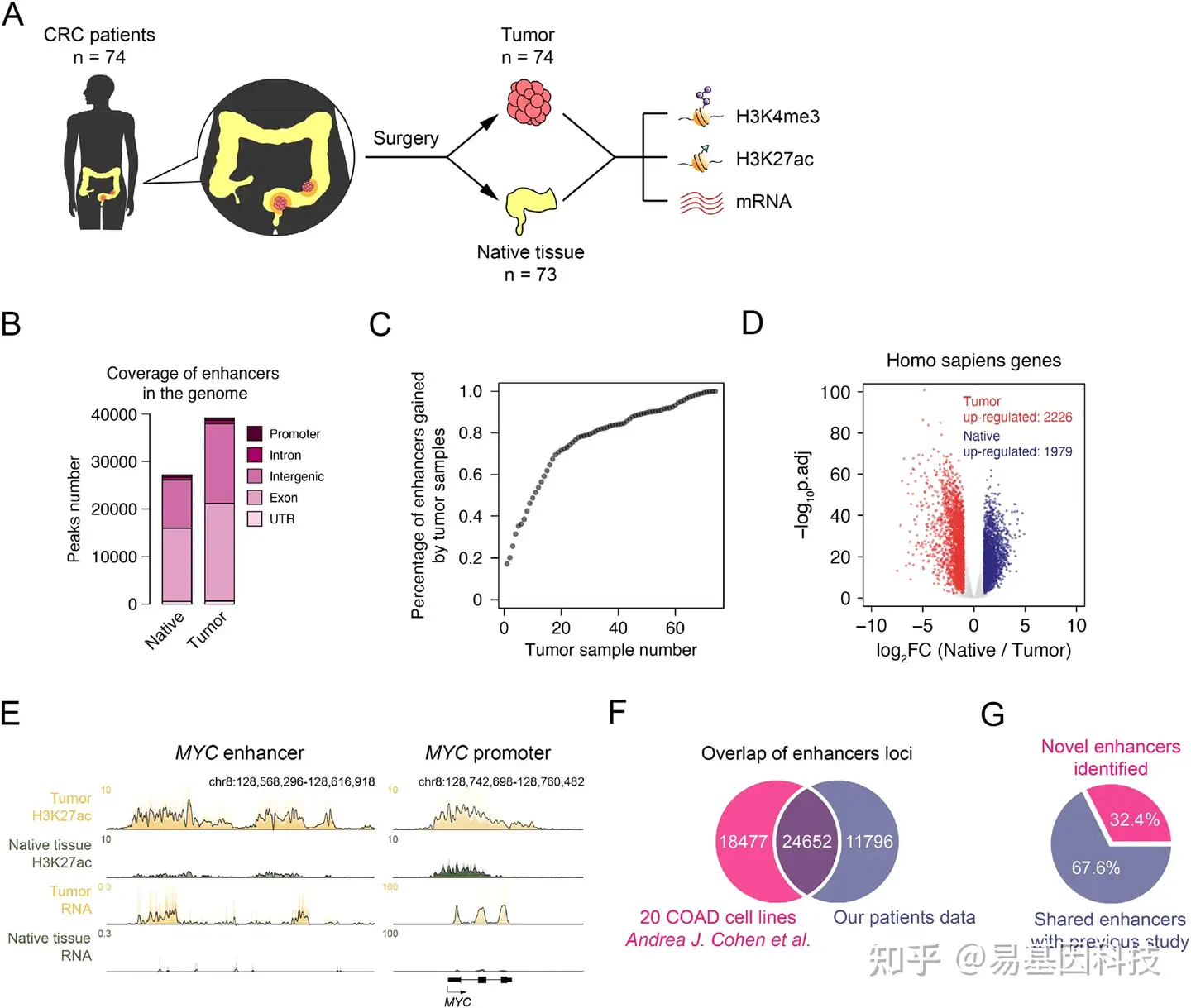
图1:CRC患者组织中活性增强子的注释
- 用于研究CRC患者的肿瘤组织和癌旁组织的增强子实验工作流程。
- CRC患者肿瘤组织和癌旁组织中增强子元件的基因组分布。
- 随着肿瘤样本数量增加,新获得的增强子占总显著增强子百分比的饱和度分析。
- 比较肿瘤和癌旁组织的基因表达倍数变化(FC)和p.adj。红点表示肿瘤上调基因,蓝点表示癌旁组织上调基因,灰点表示基因未变化。
- ChIP-seq和RNA-seq的Meta轨迹标准分析显示MYC启动子和增强子位点的H3K27ac和mRNA信号。
- 将肿瘤样本数据与20个COAD细胞系(GSE77737)之间增强子位点重叠分析。
- 本研究鉴定的CRC中新增强子的百分比。
(2)肿瘤中变化的增强子位点鉴定
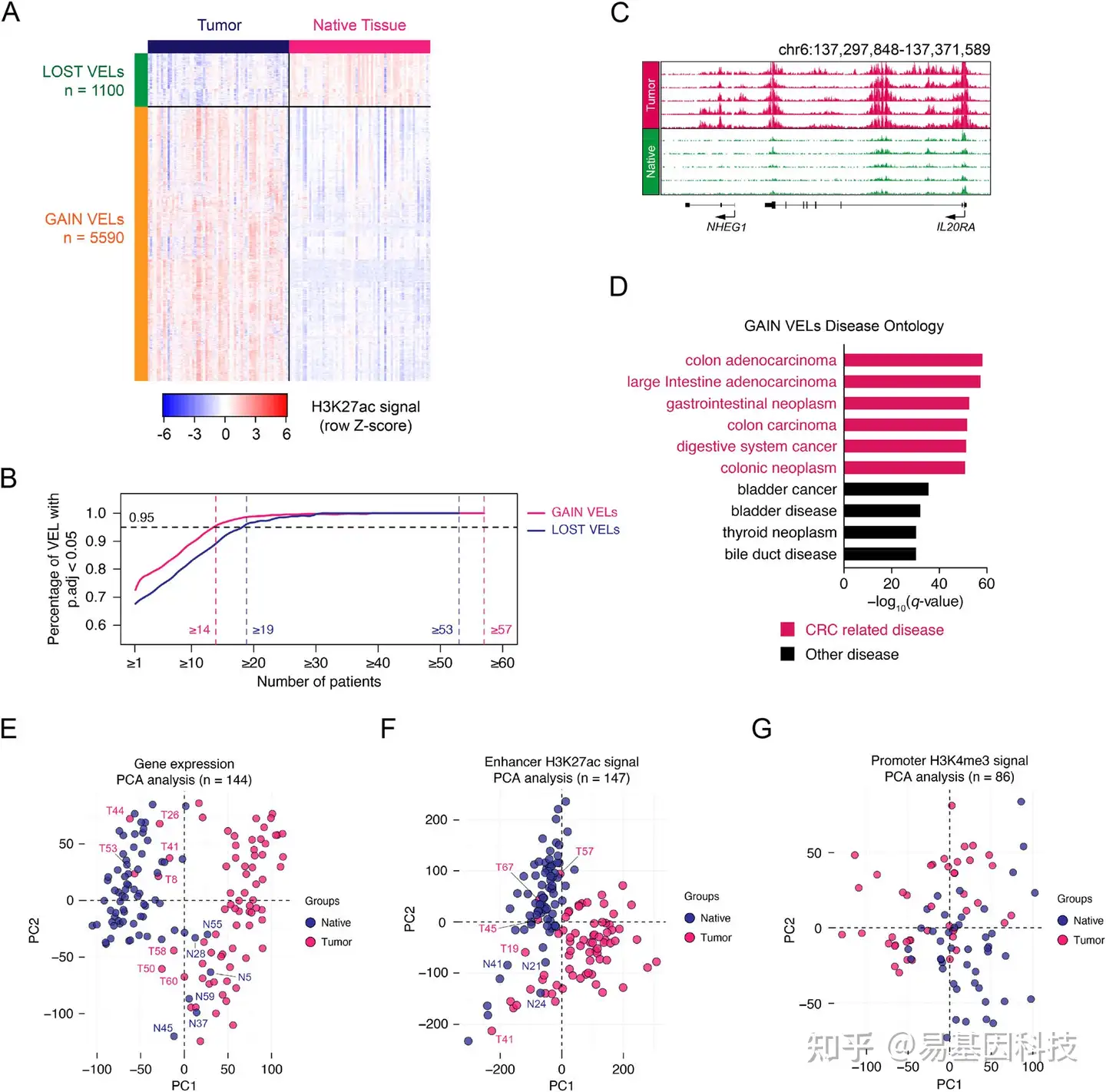
图2:CRC中增强子位点变化鉴定
- 所有肿瘤和癌旁组织中缺失和获得差异增强子位点(gain VELs)的相对H3K27ac信号。
- 达到统计学显著性的获得和缺失VEL所需的复发率(p.adj<0.05)。左侧两条垂直虚线突出显示当达到显著百分比的截止值(0.95,黑色虚线)时获得和缺失VEL的复发百分比,右侧两条线分别突出显示肿瘤或癌旁组织中获得或缺失VEL的最高复发率。gain VEL,复发达到14时,显著VEL百分比为95.635%;lost VEL,当复发达到19时,显著VEL百分比为96.273%。p.adj表示BH调整的t检验p值。
- IL20RA位点上代表性获得的差异增强子位点(gain VEL)H3K27ac轨迹。
- GREAT软件(版本3.0.0)检测的参与的人类疾病本体论分析。红色条代表CRC相关疾病,黑色条代表其他疾病。
E-G. 对RNA-seq和ChIP-seq数据鉴定的基因表达(E)、所有显著增强子(F)和启动子(G)信息对肿瘤和癌旁组织分类的PCA分析。
(3)CRC亚群的增强子特征
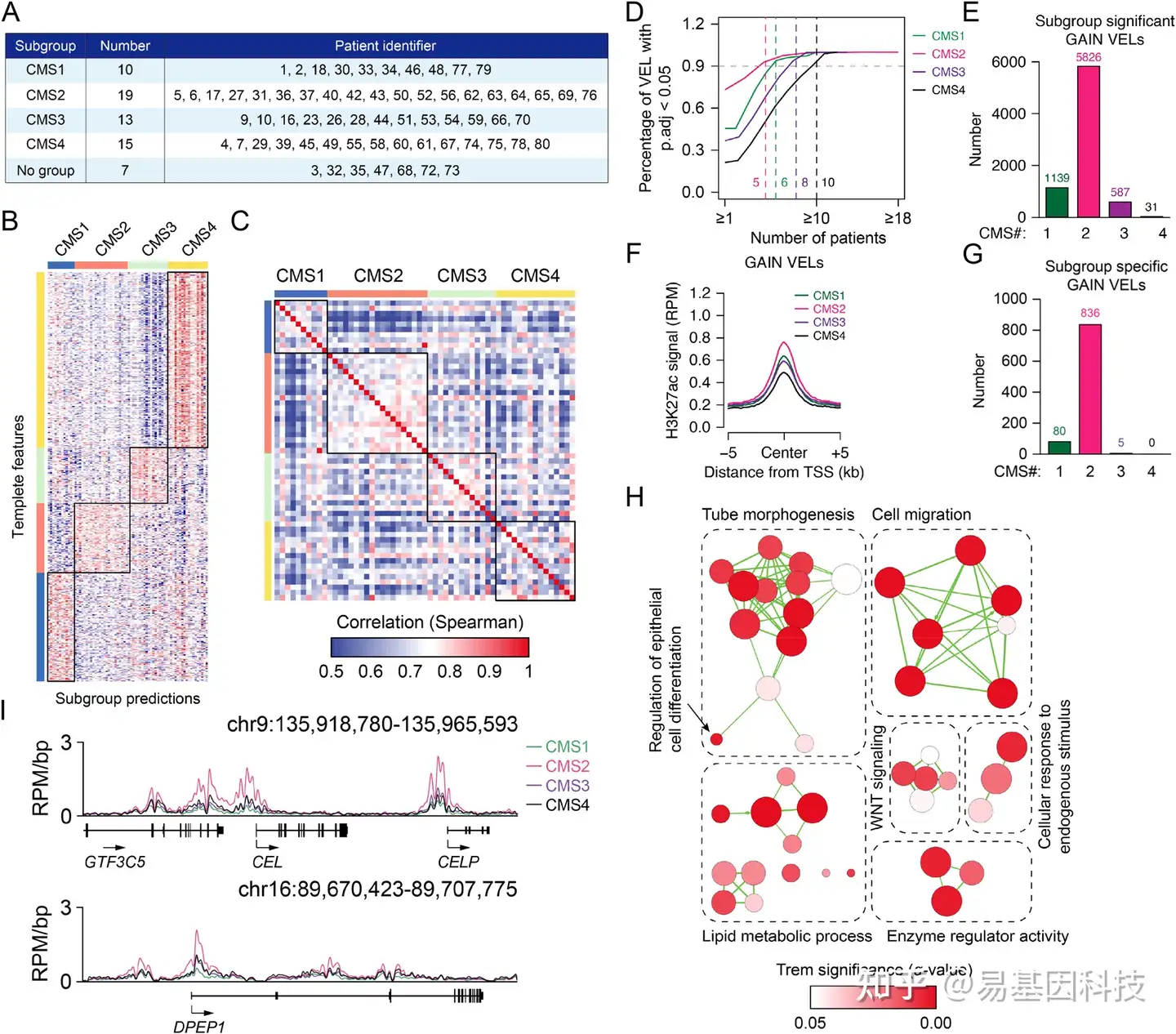
图3:CMS亚群中增强子的特征
- 四个CMS(consensus molecular subtypes)亚群成员的患者标识符。
- 使用R包CMScaller对CRC样本进行CMS分类。
- CMS1-4亚组所有肿瘤样本中gain VEL区域与H3K27ac信号相关性。相关性通过Spearman相关系数计算。
- CMS亚群中gain VEL的所需复发率,以满足不同截止值下的统计显著性(p.adj<0.05)。
- 四个CMS亚群中亚组显著gain VEL的数量。
- 四个CMS亚群中gain VEL区域上的平均H3K27ac信号(RPM)。
- 每个CMS亚群特异性gain VEL的数量。当一个CMS亚组中一个VEL的平均RPM比其他三个高1.5倍时,确定亚组特异性gain VEL。
- 基于GO(生物过程)和通路数据库(Reactome)中注释基因集的显著重叠分析,对与CMS2特异性gain VEL相关的靶基因进行功能注释。
- 四个CMS亚群中CEL和DPEP1基因位点归一化H3K27ac的Meta轨迹。
(4)超级增强子位点的分析与验证
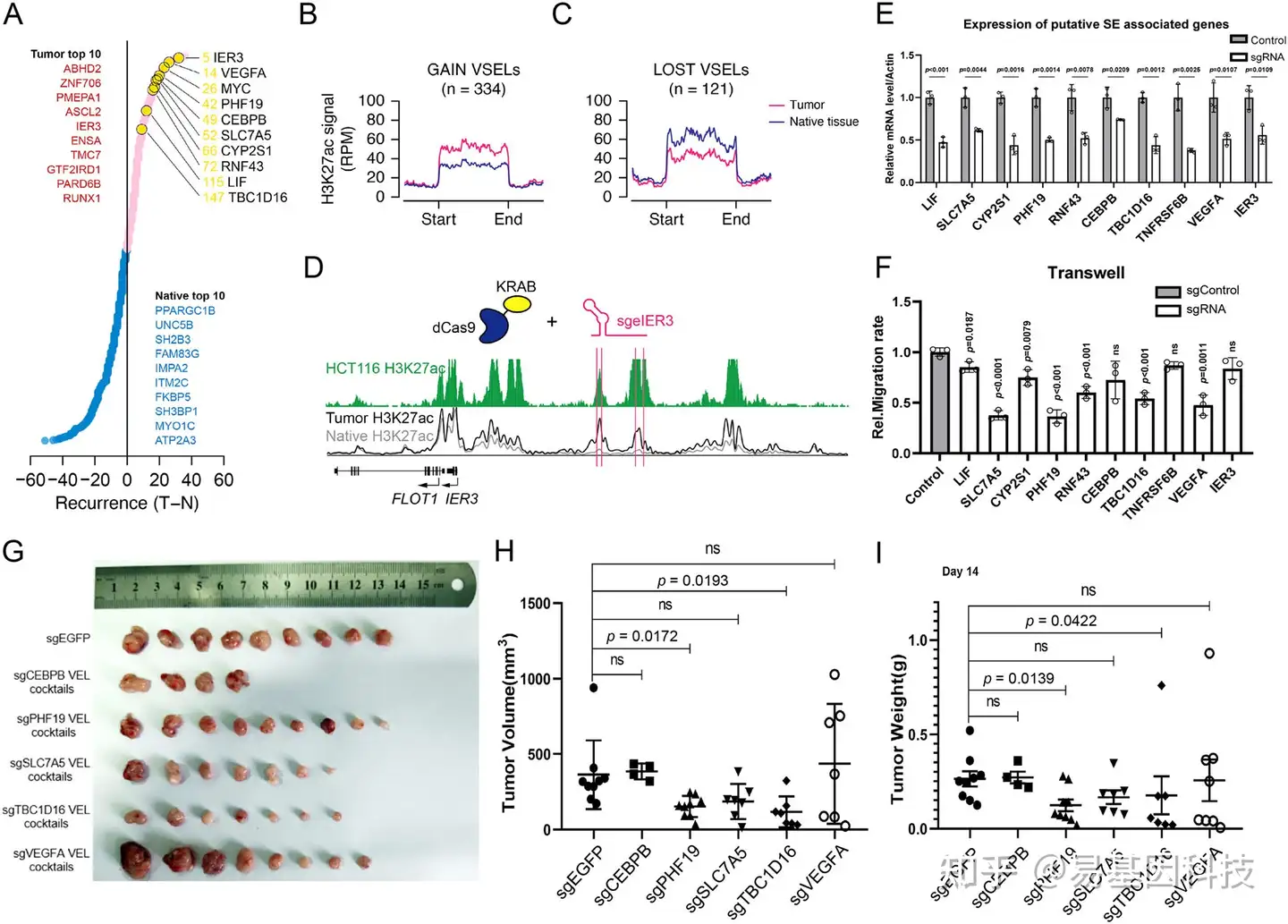
图4:肿瘤特异性超级增强子在CRC中的作用
- 与超级增强子(SE)相关的基因按复发率排序。红点代表肿瘤特异性SE基因,蓝点代表癌旁组织特异性SE基因。列出了前10个肿瘤和癌旁组织特异性基因。
- 在肿瘤和癌旁组织中获得的差异超级增强子位点(VSEL)的平均H3K27ac信号(RPM)。
- 在肿瘤和癌旁组织中缺失的差异超级增强子位点(VSEL)的平均H3K27ac信号(RPM)。
- IER3基因位点的Meta归一化H3K27ac轨迹。顶部的绿色轨迹表示HCT116中的H3K27ac信号,底部的黑色和灰色线分别表示肿瘤和癌旁组织的平均信号。粉红线表示dCas9-KRAB sgRNA靶点。
- 对照组和sgRNA组中LIF、SLC7A5、CYP2S1、PHF19、RNF43、CEBPB、TBC1D16、TNFRSF6B、VEGFA和IER3的相对mRNA水平(n=3)条形图,靶向EGFP的sgRNA对照在以下实验中用作对照*p<0.05。
- 用图5E中提到的增强子Cas9-KRAB sgRNA稳定转染的HCT116细胞系的Transwell分析(n=3)。用表达所示sgRNA的HCT116稳定细胞在裸鼠中进行G-I异种移植实验。肿瘤图像(G)、体积(H)和重量(I)。n=9(所有组别)。数据表示为平均值± SEM。使用双侧Student t检验进行统计分析。p值标记在相应项目上。
(5)功能转录因子的预测与验证
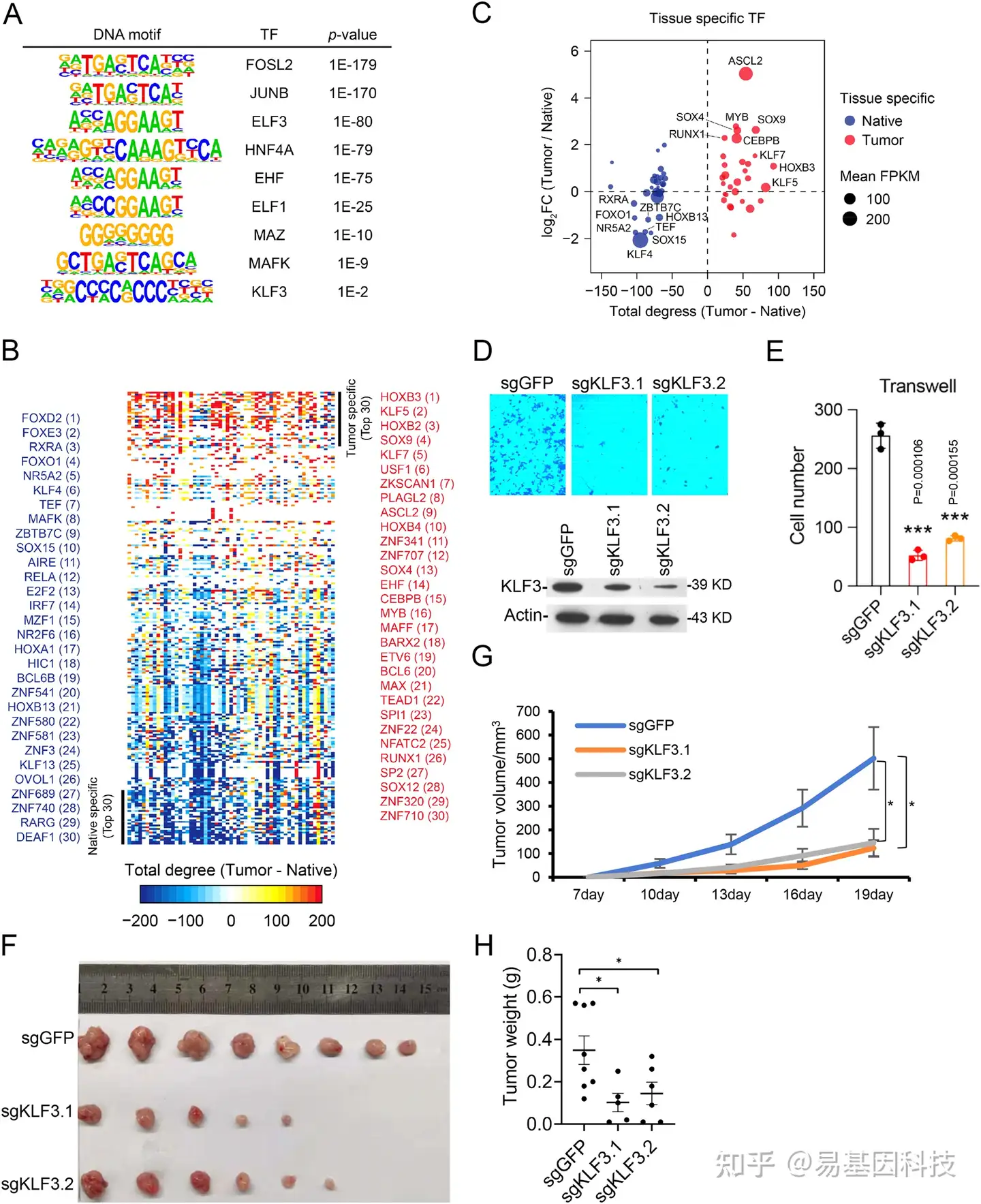
图5:CRC中功能转录因子的预测
- 通过HOMER motif分析确定在肿瘤获得水平的无核小体区域(NFR)内富集的DNA motif。
- 通过预测的核心调控回路分析肿瘤-癌旁组织的总程度(total degrees)排序转录因子热图。列出了前30个肿瘤和癌旁组织特异性TF。
- 图5B中列出的特定TF的总程度(肿瘤-癌旁组织)和表达FC(肿瘤/癌旁组织)。蓝点代表前30个肿瘤特异性TF,红点代表前30个癌旁组织特异性TF。圆圈大小表示TF在其特定组织中的平均表达(FPKM)。
D-E. KLF3敲除的HCT116细胞系Transwell分析。western blot检测KLF3,p=1.06E-4(sgKLF3.1)和1.55E-4(sgKLF3.2)。n=3,数据表示为平均值±SEM。
F-H. 将KLF3通过sgRNA稳定地敲除HCT116细胞注射到裸小鼠(10^6细胞梨小鼠,n=10),肿瘤图像(F)、肿瘤生长曲线(G)和体重(H)显示为平均值(± SEM)。G中sgKLF3.1和KLF3.2的P值分别为0.0261和0.0296,H中sgKLF3.1和KLF3.2的P值分别为0.0256和0.0355。
结论:
本研究通过对结直肠癌组织(肿瘤组织和癌旁组织)进行全基因组水平上的多组学测序(ChIP-seq、RNA-seq、全基因组测序),提供了结直肠癌临床组织的全面的活性增强子图谱,通过实验鉴定出10多个超级增强子在结直肠癌中的作用,发现调控PHF19和TBC1D16的超级增强子是致癌的超级增强子,揭示KLF3是一个新的结直肠癌的致癌转录因子,为结直肠癌的研究提供了重要的表观基因组数据和新的关键调控因子。
关于易基因染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用
- 复制因子和 DNA 修复蛋白
- 组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;
- 微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;
- 方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
有ChIP-seq测序或组学研究需要的老师可联系易基因。
参考文献:
- Li QL, et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat Commun. 2021 Nov 4;12(1):6407.
- doi:10.11843/j.issn.0366-6964.2021.012.002
- 武汉大学官网
相关阅读:
项目文章 | 组蛋白ChIP-seq揭示烟粉虱共生菌Hamiltonella调控宿主生殖新机制
项目文章 | ChIP-seq揭示HIV-1感染细胞转录抑制因子Schlafen 5的表观遗传调控机制
项目文章|ChIP-seq揭示H3K27me3去甲基化酶在体细胞重编程调控转录机制

标签:转录,seq,Nature,组学,ChIP,癌旁,增强子,肿瘤,位点 From: https://www.cnblogs.com/E-GENE/p/17147319.html