1. 背景
在前面的教程中,我们从数据集中删除了低质量的细胞,包括计数较差以及双细胞,并将数据存放在anndata文件中。由于单细胞测序技术的限制,我们在样本中获得RNA的时候,经过了分子捕获,逆转录还有测序。这些步骤会影响同一种细胞的细胞间的测序计数深度的变异性,故单细胞测序数据中的细胞间差异可能会包含了这部分测序误差,等价于计数矩阵中包含了变化很大的方差项。但在目前的统计方法中,绝大部分模型都预先假定了数据具有相同的方差结构。
伽玛-柏松分布
从理论上和经验上建立的 UMI 数据模型是 Gamma-Poisson 分布,即\(Var[Y] = \mu + \alpha \mu^2\),其中\(\mu\)代表UMI平均值,\(\alpha\)代表细胞UMI的过度离散值。若 \(\alpha=0\) 时,意味着此时UMI的分布为泊松分布。
“归一化”的预处理步骤旨在通过将“UMI的方差”缩放到指定范围,来调整数据集中的原始UMI计数以实现模型建模。而在真实的单细胞分析中,有不同的归一化方法以解决不同的分析问题。但经验发现,移位对数在大部分数据中的表现良好,这在2023年4月的Nature Method上的基准测试中有提到。
本章将向读者介绍两种不同的归一化技术:移位对数变换和皮尔逊残差的解析近似。移位对数有利于稳定方差,以利于后续降维和差异表达基因的识别。皮尔森近似残差可以保留生物学差异,并鉴定稀有细胞类型。
我们首先导入我们所需要的Python包,以及上一个教程分析所得到的anndata文件。
import omicverse as ov
import scanpy as sc
ov.utils.ov_plot_set()
adata = sc.read(
filename="s4d8_quality_control.h5ad",
backup_url="https://figshare.com/ndownloader/files/40014331",
)
try downloading from url
https://figshare.com/ndownloader/files/40014331
... this may take a while but only happens once
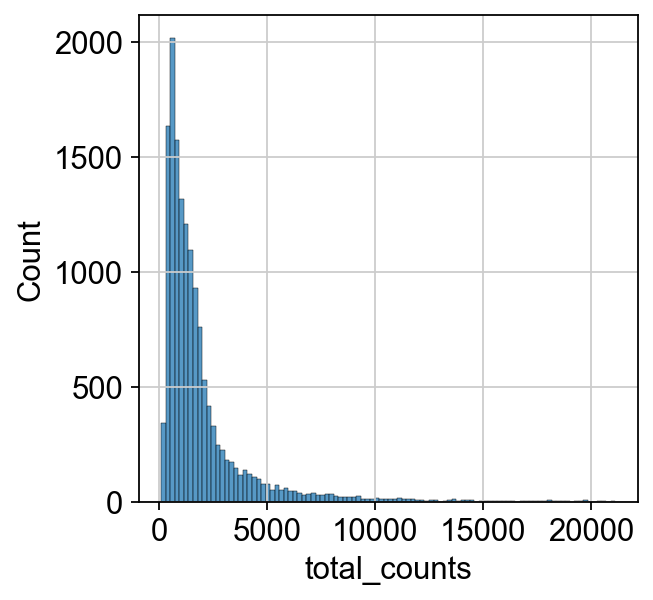
我们首先检查原始计数UMI的分布,一般在后续的分析中我们会忽略这一步,但对该分布的认识有利于我们理解归一化的意义。
import seaborn as sns
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False)
2. 移位对数
我们将介绍的第一个归一化方法是基于delta方法的移位对数。Delta方法应用非线性函数\(f(Y)\),使得原始计数 \(Y\) 中的差异更加相似。
我们定义非线性函数\(f(Y)\)的变换如下:
\[f(y) = \log(\frac{y}{s}+y_0) \]其中\(y\)是原始的计数,\(s\)是尺寸因子,\(y_0\)是伪计数。我们为每一个细胞确定自己的尺寸因子,以满足同时考虑采样效果和不同细胞尺寸的变换。细胞的尺寸因子可以计算为:
\[s_c = \frac{\sum_g y_{gc}}{L} \]其中 \(g\)代表不同的基因,\(L\)代表基因的计数总和。确定尺寸因子的方法有很多,在scanpy中,我们默认使用原始计数深度的中位数来计算,而在seruat中使用固定值\(L=10^4\),而在omicverse的预处理中,我们将\(L\)设定为\(50*10^4\)。不同的值会使得过度离散值 \(\alpha\)的不同。
过度离散值 \(\alpha\)
过度离散值 \(\alpha\)描述了数据集中存在着比期望更大的变异性。
移位对数是一种快速归一化技术,优于其他揭示数据集潜在结构的方法(特别是在进行主成分分析时),并且有利于方差的稳定性,以进行后续的降维和差异表达基因的识别。我们现在将检查如何将此归一化方法应用于我们的数据集。我们可以使用pp.normalized_total来使用 scanpy 调用移位对数。并且我们设置target_sum=None,inplace=False来探索两种不同的归一化技术。
scales_counts = sc.pp.normalize_total(adata, target_sum=None, inplace=False)
# log1p transform
adata.layers["log1p_norm"] = sc.pp.log1p(scales_counts["X"], copy=True)
normalizing counts per cell
finished (0:00:00)
我们使用hist图来对比归一化前后的计数变化
import matplotlib.pyplot as plt
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(adata.layers["log1p_norm"].sum(1), bins=100, kde=False, ax=axes[1])
axes[1].set_title("Shifted logarithm")
plt.show()
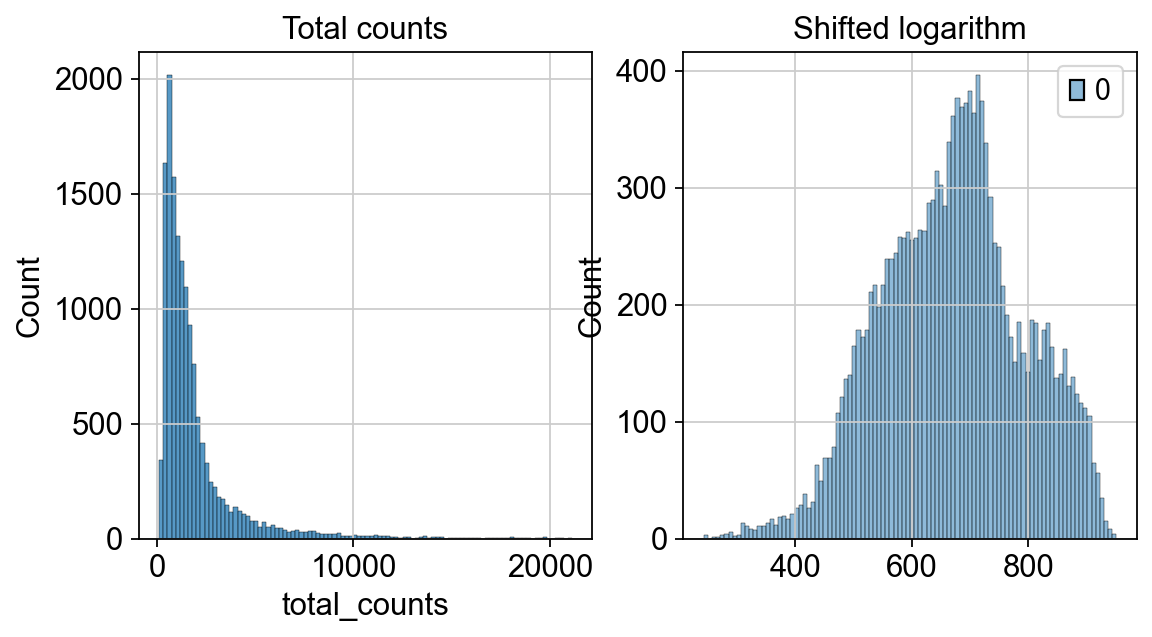
我们发现nUMI的最大值在1000左右,经过移位对数化后,并且移位对数化后的nUMI的分布近似正态分布。
3. 皮尔森近似残差
我们观察到,scRNA-seq数据中的细胞间变异包括了生物异质性以及技术效应。而移位计数并不能很好的排除两种不同变异来源的混淆误差。皮尔森近似残差利用了“正则化负二项式回归”的皮尔森残差来计算数据中潜在的技术噪音,将计数深度添加为广义线性模型中的协变量,而在不同的归一化方法的测试中,皮尔森残差法可以消除计数效应带来的误差,并且保留了数据集中的细胞异质性。
此外,皮尔森残差法不需要进行启发式步骤,如伪计数加法/对数变化,该方法的输出就是归一化后的值,包括了正值和负值。细胞和基因的负残差表明与基因的平均表达和细胞测序深度相比,观察到的计数少于预期。正残差分别表示计数越多。解析 Pearon 残差在 scanpy 中实现,可以直接在原始计数矩阵上计算。
from scipy.sparse import csr_matrix
analytic_pearson = sc.experimental.pp.normalize_pearson_residuals(adata, inplace=False)
adata.layers["analytic_pearson_residuals"] = csr_matrix(analytic_pearson["X"])
computing analytic Pearson residuals on adata.X
finished (0:00:15)
fig, axes = plt.subplots(1, 2, figsize=(8, 4))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(
adata.layers["analytic_pearson_residuals"].sum(1), bins=100, kde=False, ax=axes[1]
)
axes[1].set_title("Analytic Pearson residuals")
plt.show()
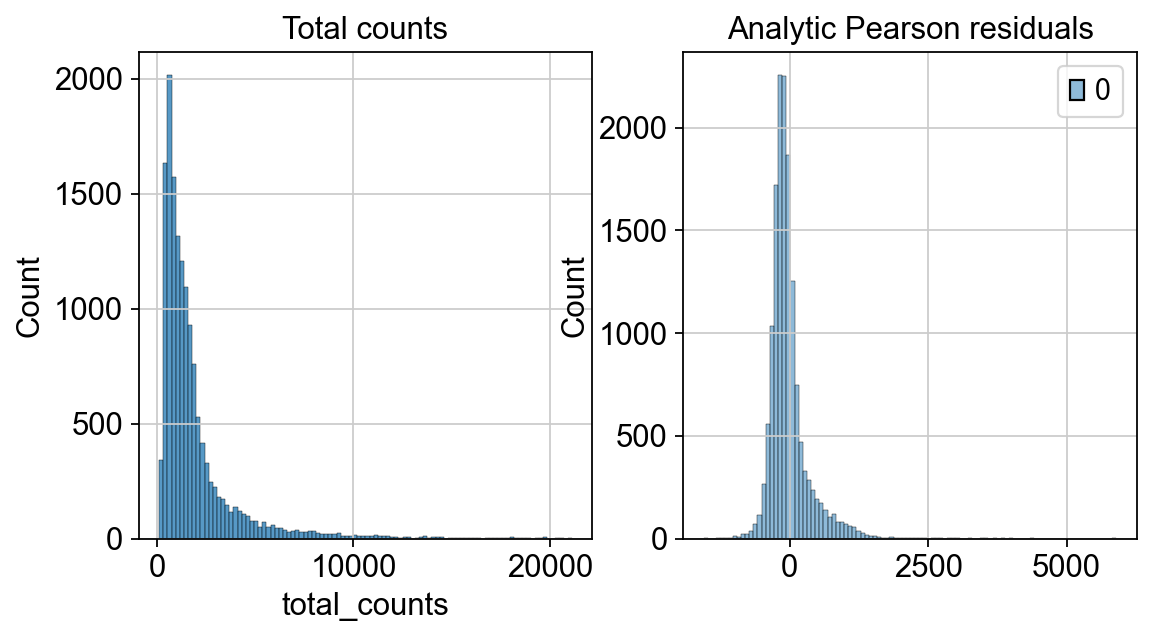
如果我们设置inplace=False时,我们归一化的计数矩阵会取代原anndata文件中的计数矩阵,即更改adata.X的内容。
4. 一键式归一化
我们在omicverse中提供了预处理函数pp.preprocess,该方法可直接计算移位对数或皮尔森残差,方法内同时包括了基于移位对数/皮尔森残差的高可变基因的选择方法,高可变基因会在下一节的教程中进行讲解。
此外,我们omicverse的归一化方法还提供了稳定基因的识别与过滤。
4.1 移位对数
在omicverse中,我们设置mode='shiftlog|pearson'即可完成移位对数的计算,一般来说,默认的target_sum=50*1e4,同时高可变基因定义为前2000个,需要注意的是,当omicverse的版本小于1.4.13时,mode的参数只能设置为scanpy或pearson,scanpy与shiftlog的意义是相同的
adata_log=ov.pp.preprocess(adata,mode='shiftlog|pearson',n_HVGs=2000,)
adata_log
Begin log-normalization, HVGs identification
After filtration, 20171/20171 genes are kept. Among 20171 genes, 20171 genes are robust.
End of log-normalization, HVGs identification.
Begin size normalization and pegasus batch aware HVGs selection or Perason residuals workflow
normalizing counts per cell The following highly-expressed genes are not considered during normalization factor computation:
[]
finished (0:00:00)
2000 highly variable features have been selected.
End of size normalization and pegasus batch aware HVGs selection or Perason residuals workflow.
4.2 皮尔森近似残差
我们也可以设置mode='pearson|pearson'来完成皮尔森近似残差的计算,此时我们不需要输入target_sum,需要注意的是,当omicverse的版本小于1.4.13时,mode的参数只能设置为scanpy或pearson
adata_pearson=ov.pp.preprocess(adata,mode='pearson|pearson',n_HVGs=2000,)
adata_pearson
Begin log-normalization, HVGs identification
After filtration, 20171/20171 genes are kept. Among 20171 genes, 20171 genes are robust.
End of log-normalization, HVGs identification.
Begin size normalization and pegasus batch aware HVGs selection or Perason residuals workflow
extracting highly variable genes
--> added
'highly_variable', boolean vector (adata.var)
'highly_variable_rank', float vector (adata.var)
'highly_variable_nbatches', int vector (adata.var)
'highly_variable_intersection', boolean vector (adata.var)
'means', float vector (adata.var)
'variances', float vector (adata.var)
'residual_variances', float vector (adata.var)
computing analytic Pearson residuals on adata.X
finished (0:00:04)
End of size normalization and pegasus batch aware HVGs selection or Perason residuals workflow.
我们对比一下两种分析的结果
import matplotlib.pyplot as plt
import seaborn as sns
fig, axes = plt.subplots(1, 3, figsize=(12, 4))
p1 = sns.histplot(adata.obs["total_counts"], bins=100, kde=False, ax=axes[0])
axes[0].set_title("Total counts")
p2 = sns.histplot(
adata_log.X.sum(1), bins=100, kde=False, ax=axes[1]
)
axes[1].set_title("Shifted logarithm")
p3 = sns.histplot(
adata_pearson.X.sum(1), bins=100, kde=False, ax=axes[2]
)
axes[2].set_title("Analytic Pearson residuals")
plt.show()
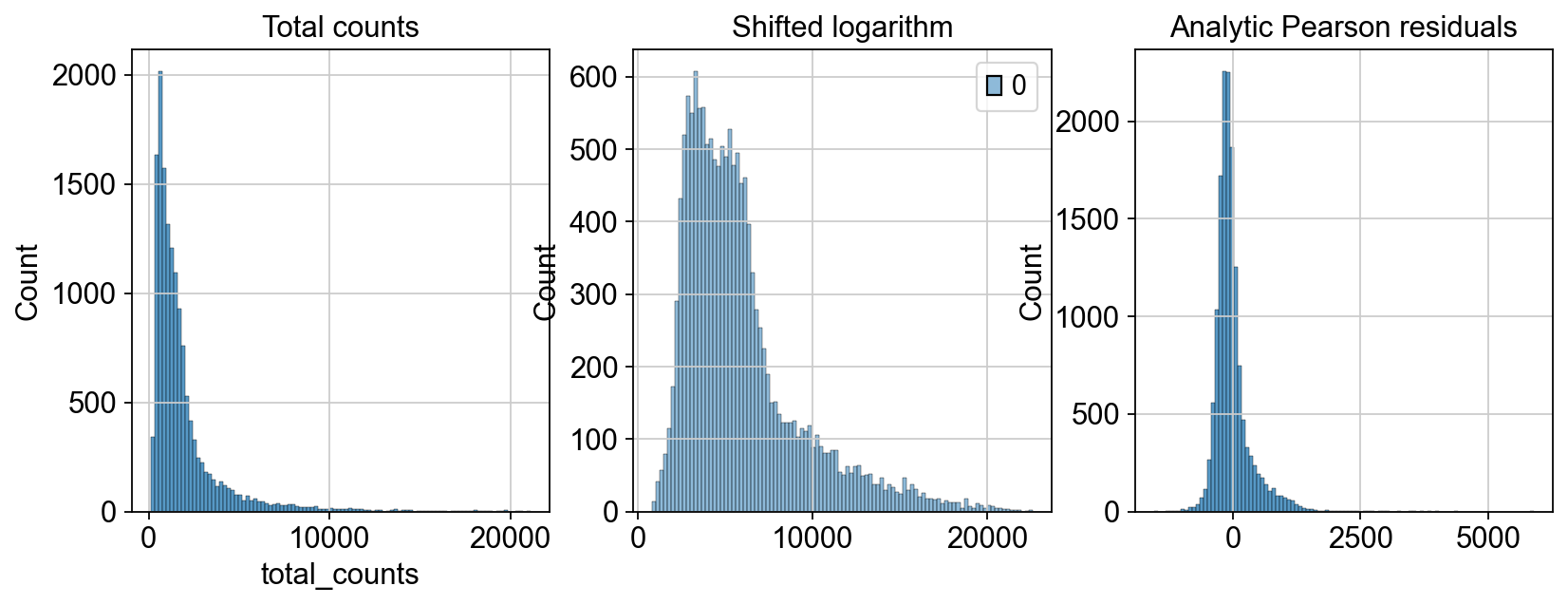
以上,就是本章所要介绍的归一化内容,通过benchmark的测试,我们发现移位对数适用于大多数任务。但是如果我们的分析目标是寻找稀有细胞的时候,可以考虑采用皮尔森残差法来进行归一化。
5. 思考
为了加深你对本章的理解,我们提出了以下思考题,如有兴趣作答者,可将答案发送至邮箱starlitnightly@163.com,邮件标题为姓名/昵称-单细胞最好教程(二)思考题
- 我们在进行移位对数分析的时候,
sc.pp.normalize_total(adata, target_sum=None, inplace=False)中的target_sum使用了默认值,在seurat`中默认值是10,000,在一些教程中设定为1,000,000,我们虽然对这个值的意义进行了简单介绍,但你认为不同的值背后的含义是什么? - 我们为什么会使用皮尔森残差来计算归一化值,相对于移位对数而言有什么更好的地方?
- 你可以找出别的归一化方法,并比较其与移位对数,皮尔森残差的好坏吗?
如果你对omicverse的使用有什么疑问,或者也想加入一起开发擅长,想在生物信息学工具开发路上找到同道中人,那么这个Python的转录组学分析框架与生态交流群请不要错过。感谢Jimmy老师对omicverse的大力支持。
如果群满了也可以关注我们的github仓库获取最新消息:https://github.com/Starlitnightly/omicverse
标签:教程,pearson,axes,残差,计数,单细胞,归一化,adata From: https://www.cnblogs.com/starlitnightly/p/18258580