今天处理一个客户的基因型数据,遇到了一个格式转化的问题。
我想将标准vcf转为某特定格式(类hapmap),之前基于pysam库写了一个比较成熟的脚本,运行了n年都没问题。
但这次出错如下:
a,b = v['GT']
ValueError: not enough values to unpack (expected 2, got 1)
如果你直接去网上搜索该错误,会有一堆教程告诉你是因为文件中有多余的换行符或者编码错误,但事实并非如此。不如直接定位脚本,发现pysam中samples.iteritems方法错误,GT应该有两个等位基因才对。
for sample,v in rec.samples.iteritems():
a,b = v['GT']
if a == None or b == None:
gt = '-'
else:
gt = ''.join([alleles[a],alleles[b]])
gts.append(gt)
h.extend(gts)
根据结果,我debug到开始出错的那个位点,发现GT为“.”,而非我们常见的“./.”或“.|.”,因此pysam解析不了。但大多数缺失是我们常见的形式,因此我不明白为什么会出现这种情况。
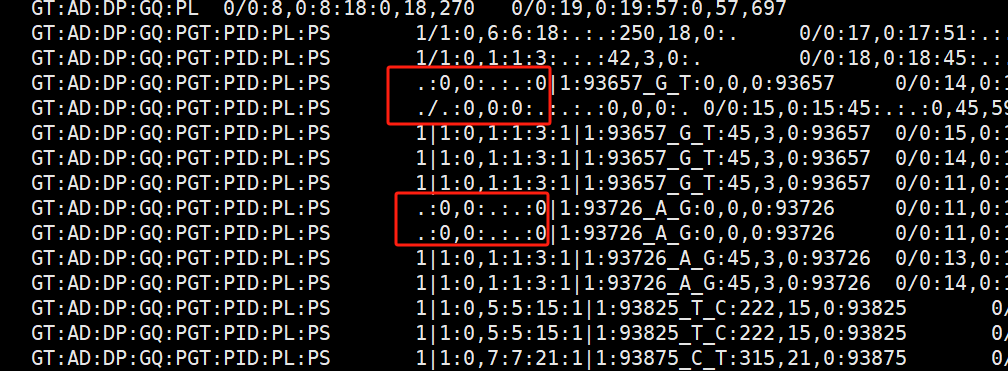
网上查了下,似乎没有专门针对这个问题的答案。如果有朋友知道原因,请不吝赐教。我推测可能是某些软件(比如GATK)版本的问题。
我们知道,GATK3和GATK4有比较大的区别,而从 GATK 4.2.3.0 开始又有较大改动,比如缺失值的显示变为0了,而非之前版本的点。
当然我们可以通过DP属性等于0来和REF base做区分(比如bcftools即可处理),但总归要多处理一下,不太方便的。而官方宣称这样做是为了使其运行得更快、更高效,以进行大规模的joint calling。

版本更新导致文件格式不统一,这是个很痛苦的事情。所以小编建议,在正式数据分析之前,大家最好搞清楚上游软件版本,尤其是结果差异较大版本,以免发生不必要的错误。
当然,找到了错误的地方,我的问题很容易解决,写个代码统一下缺失值表示即可。如果你遇到类似的错误不会解决,可以找我要脚本。