一起学生信分析 RNA-Seq下游 篇
DESeq2介绍
专为高通量测序数据(特别是 RNA-seq 数据)设计,用于分析计数数据的差异表达,同样功能的还有 limma 和 edgR。
差异表达分析
使用DESeq2进行差异分析
本教程使用的数据下载链接:
表达矩阵 matrix_clean.txt 下载:https://wwry.lanzouq.com/i6w7M167f9sh
metadata.txt 下载:https://wwry.lanzouq.com/iAUHM167f9ti
diffexp_result.txt 下载:https://wwry.lanzouq.com/i6w7M167f9sh
导入工作R包
library(DESeq2)
library("RColorBrewer")
suppressMessages(library("gplots"))
library("amap")
library("reshape2")
library("ggplot2")
library("ggrepel")
library("pheatmap")
suppressMessages(library(DESeq2))
library("BiocParallel")
library("tximport")
library("readr")
读取数据
# 定义输入输出文件路径
# 输入的基因表达矩阵文件为matrix_clean.txt。
input_matrix='matrix_clean.txt' #将前面得到的表达信息文件matrix.txt的绝对路径及文件写在此处的''内
# 输入的样本信息文件为metadata.txt。
input_info='metadata.txt' #将预先整理的样本信息文件sample_info.txt的绝对路径及文件写在此处的''内
# 输出文件将保存为DESeq2/diffexp_result.txt。
output_file='DESeq2/diffexp_result.txt' #将保存输出文件的绝对路径及文件写在此处的''内
##################################################################
# 数据读入与处理
input_data <- read.table(input_matrix,header = TRUE,row.names = 1)
input_data <- round(input_data,digits=0) # 读取基因表达矩阵,将其四舍五入为整数
head(input_data)
info_data <- read.table(input_info,header = TRUE,row.names = 1, com='',
quote='', check.names=F, sep="\t")
info_data
# input_data 结构如下
# A1 A2 A3 W1 W2 W3
# G4B84_000001 0 0 0 0 4 0
# G4B84_000002 0 0 0 0 0 0
# G4B84_000003 0 0 0 0 0 0
# G4B84_000004 0 0 0 0 0 0
# G4B84_000005 0 2 0 0 6 2
# G4B84_000006 0 6 2 8 2 2
# info_data 结构如下
# Group
# A1 A
# A2 A
# A3 A
# W1 W
# W2 W
# W3 W
创建DESeq对象并使用DESeq函数进行差异表达分析
# 注意这里的 design = ~Group 必须是 info_data 中的分组列名
# 使用 DESeqDataSetFromMatrix 创建一个 DESeq 数据集即 dds 对象
dds <- DESeqDataSetFromMatrix(countData = input_data, colData = info_data, design = ~Group) # 若batch为空则design=~label
# 开始进行差异表达分析
# DESeq 这个函数会基于之前定义的设计公式(在这里是基于"Group"列)来分析哪些基因在不同组之间表现出显著差异。分析完成后,结果会储存在dds对象中。
dds <- DESeq(dds)
处理DESeq2的差异表达分析结果
提取差异表达分析结果:它从dds对象中提取差异表达基因的统计结果,并考虑了一个特定的显著性阈值(alpha = 0.1)。
# results: 这是DESeq2包的一个函数, 用于从上述的dds对象中提取差异分析的结果。
# alpha=0.1 这是为结果添加了一个列来标记那些在给定的alpha值下被视为显著差异的基因。
# 需要注意的是 alpha 在 results 中的主要作用是优化独立过滤的阈值,而不是直接筛选基于padj的显著基因。
# 当然 alpha 的默认值就是 0.1,此处不写也可以
result <- results(dds,alpha = 0.1)
summary(result)
排序差异基因:它按照调整后的p值 (padj) 对差异表达的基因进行排序,这意味着最显著差异的基因会被放在结果的顶部。
result <- result[order(result$padj),]
合并归一化读数计数:它将差异表达结果与归一化的读数计数进行合并。这样,在同一个文件中,你既可以看到每个基因的差异表达统计数据,也可以看到每个基因在每个样本中的表达水平(经过归一化)。
# 获得归一化表
result_data <- merge(as.data.frame(result),as.data.frame(counts(dds,normalized=TRUE)),by='row.names',sort=FALSE)
names(result_data)[1] <- 'Gene'
write.table(result_data,file=output_file,sep = '\t',quote=F,row.names = F)
对数据进行 归一化 和 rlog变换
# Get normalized counts
print("Output normalized counts")
# 归一化读数计数
# 从dds对象中获取归一化的读数计数。这些归一化的读数是经过DESeq2内部算法调整后的,以消除样品之间的大小因子和其他潜在差异。
normalized_counts <- counts(dds, normalized=TRUE)
# 标准化的结果按整体差异大小排序
## 使用apply函数计算每个基因的中位数绝对偏差(MAD)。这为下一步提供了每个基因表达变异性的度量。
normalized_counts_mad <- apply(normalized_counts, 1, mad)
## 根据MAD值对基因进行排序,这样差异最大的基因会放在最前面。
normalized_counts <- normalized_counts[order(normalized_counts_mad, decreasing=T), ]
# 这里设置待会输出的文件的前缀,因为这里使用的黄曲霉菌所以简写 Asp
output_prefix = 'Asp'
# 常规R输出忽略左上角(输出文件第一列也就是基因列的列名字)
# 输出结果为完整矩阵,保留左上角的id。
normalized_counts_output = data.frame(id=rownames(normalized_counts), normalized_counts)
write.table(normalized_counts_output, file=paste0(output_prefix,".DESeq2.normalized.xls"),
quote=F, sep="\t", row.names=F, col.names=T)
# rlog变换
## 使用DESeq2的rlog变换对读数进行对数变换。rlog变换是对数据进行稳健的对数变换,有助于使数据更加正态分布。
rld <- rlog(dds, blind=FALSE)
## 从rld对象中提取变换后的读数矩阵。
rlogMat <- assay(rld)
## 根据前面计算的MAD值对rlog变换后的读数进行排序。
rlogMat <- rlogMat[order(normalized_counts_mad, decreasing=T), ]
## 保存 rlogMat
print("Output rlog transformed normalized counts")
rlogMat_output = data.frame(id=rownames(rlogMat), rlogMat)
write.table(rlogMat_output, file=paste0(output_prefix,".DESeq2.normalized.rlog.xls"),
quote=F, sep="\t", row.names=F, col.names=T)
画图
- 小提琴箱线图
##################################
## 小提琴图
##################################
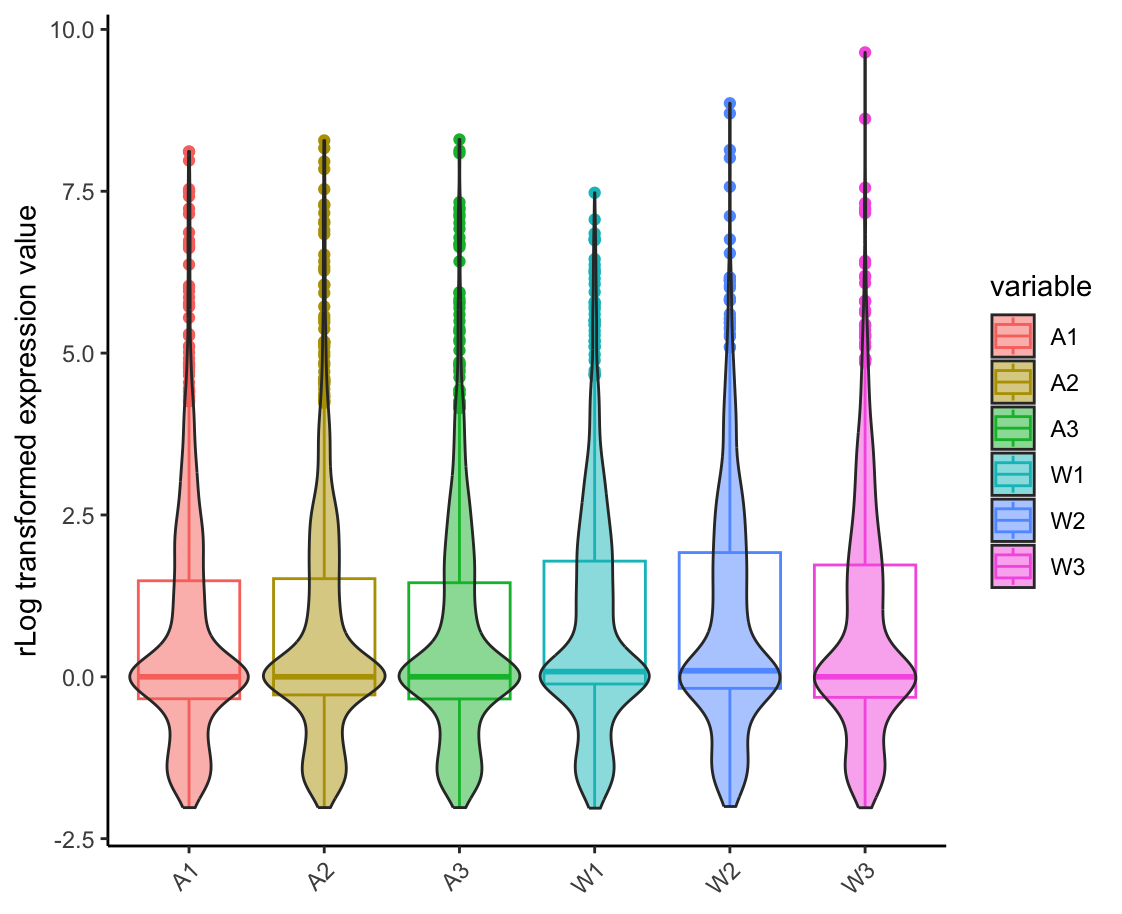
## 标准化后样品的表达分布
rlog_mat_melt <- melt(rlogMat_output, id.vars = c('id'))
ggplot(rlog_mat_melt, aes(x=variable, y=value)) + geom_boxplot(aes(color=variable)) +
geom_violin(aes(fill=variable), alpha=0.5) + theme_classic() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1),
axis.title.x = element_blank()) + ylab("rLog transformed expression value")
ggsave(filename = paste0(output_prefix, ".DESeq2.normalized.rlog.violin.pdf"), width=13.5,height=15,units=c("cm"))
- Pearson图
## 样本层级聚类
##################################
## pearson图
##################################
print("Performing sample clustering")
hmcol <- colorRampPalette(brewer.pal(9, "GnBu"))(100)
# 使用cor函数计算rlogMat的Pearson相关系数,并将其四舍五入到四位小数
pearson_cor <- round(as.matrix(cor(rlogMat, method="pearson")),4)
# 用的是 pearson
# 使用hcluster函数对rlogMat进行层级聚类,计算方法选择为"pearson"。
hc <- hcluster(t(rlogMat), method="pearson")
# 从给定的相关性矩阵中提取其下三角部分和上三角部分,分别将其它的部分设为NA。
# Get lower triangle of the correlation matrix
get_lower_tri<-function(cormat){
cormat[upper.tri(cormat)] <- NA
return(cormat)
}
# Get upper triangle of the correlation matrix
get_upper_tri <- function(cormat){
cormat[lower.tri(cormat)]<- NA
return(cormat)
}
# pearson_cor被重新排序,以匹配层级聚类的结果顺序。
pearson_cor <- pearson_cor[hc$order, hc$order]
pearson_cor_output = data.frame(id=rownames(pearson_cor), pearson_cor)
# 保存排序后的Pearson相关系数矩阵:
# 拿到pearson的数据后可以做图
write.table(pearson_cor_output, file=paste0(output_prefix,".DESeq2.pearson_cor.xls"),
quote=F, sep="\t", row.names=F, col.names=T)
upper_tri <- get_upper_tri(pearson_cor)
# Melt the correlation matrix
melted_cormat <- melt(upper_tri, na.rm = TRUE)
col = colorRampPalette(rev(brewer.pal(n=7, name="RdYlBu")))(100)
# Create a ggheatmap
p <- ggplot(melted_cormat, aes(Var2, Var1, fill = value)) +
geom_tile(color = "white") +
scale_fill_gradientn(colours = col, name = "Pearson correlation") +
theme_classic() +
coord_fixed() +
theme(
axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1),
axis.title.x = element_blank(),
axis.title.y = element_blank(),
legend.justification = c(1, 0),
legend.position = "top",
legend.direction = "horizontal"
) +
guides(fill = guide_colorbar(
barwidth = 8,
barheight = 1,
title.position = "left"
))
p
ggsave(filename=paste0(output_prefix,".DESeq2.normalized.rlog.pearson.pdf"),width=13.5,height=15,units=c("cm"))
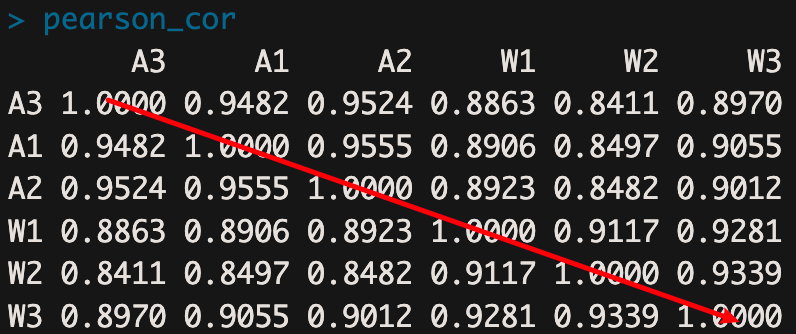
pearson_cor的结构如下图:我们可以看到其是对称矩阵,所以get_lower_tri和get_upper_tri这两个函数是提取矩阵的下三角和上三角部分
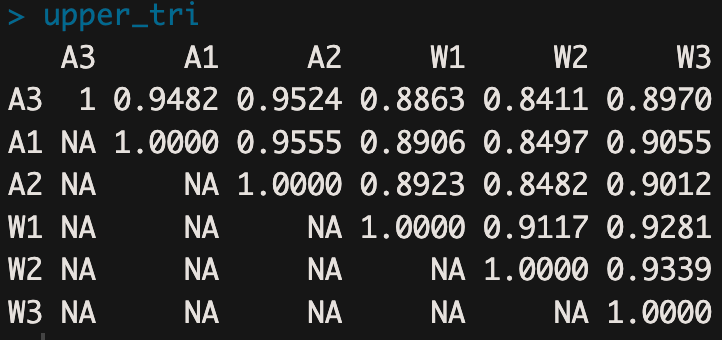
我们可以查看一下upper_tri这个数据的结构,可以看到取得是上三角数据,其对称数据置为 NA
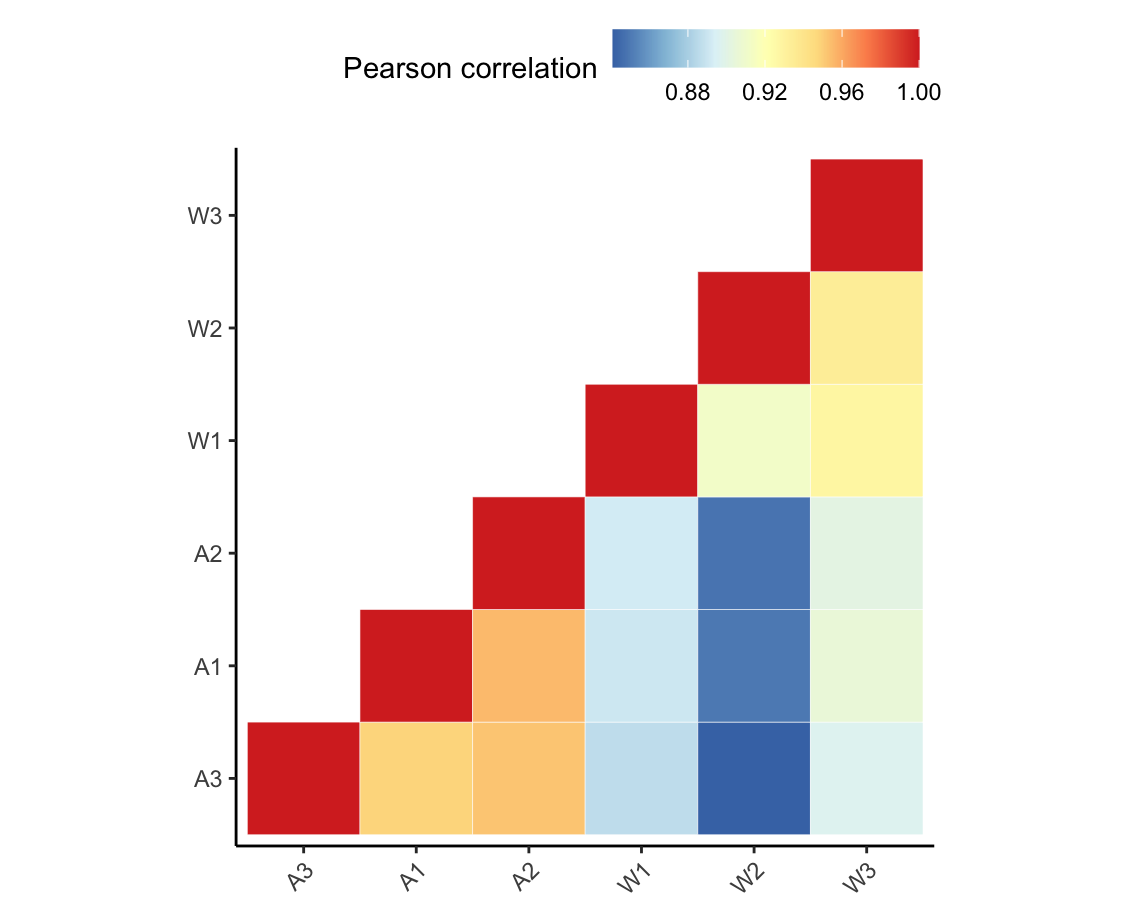
绘制的热图结果:
- PCA图
## PCA聚类分析
##################################
## PCA图
##################################
# 对正则化对数变换(rlog)后的表达数据 执行 主成分分析(PCA)
formulaV <- c("Group")
# 选择前500行进行PCA。
topn = 500
rlogMat_nrow = nrow(rlogMat)
# 如果rlogMat中的行数小于500,就使用所有行。
if (topn > rlogMat_nrow){
topn = rlogMat_nrow
}
# 为接下来的PCA准备数据
# 选择前topn行的数据,然后将数据转置,使样本成为行,基因为列。
pca_mat = rlogMat[1:topn,]
pca_mat <- as.data.frame(t(pca_mat))
# 执行PCA,scale=T代表数据会被标准化
pca <- prcomp(pca_mat, scale=T)
# pca结果提取主成分
pca_x = pca$x
sample = info_data
# 保存PCA结果
pca_individual = data.frame(samp=rownames(pca_x), pca_x, sample)
write.table(pca_individual, file=paste0(output_prefix,".DESeq2.pca_individuals.xls"), sep="\t", quote=F, row.names=F, col.names=T)
# 计算PCA的方差解释度
pca_percentvar <- formatC(pca$sdev^2 * 100 / sum( pca$sdev^2))
# 根据formulaV, 如果formulaV只包含一个元素(即只有一个分组变量),则只使用颜色来表示样本的组别。
# 如果formulaV有两个元素,则既使用颜色也使用形状来表示样本的组别
if (length(formulaV)==1) {
p <- ggplot(pca_individual, aes(PC1, PC2, color=Group))
} else if (length(formulaV==2)) {
p <- ggplot(pca_data, aes(PC1, PC2, color=Group,
shape=Group))
}
p = p + geom_point(size=3) +
xlab(paste0("PC1: ", pca_percentvar[1], "% variance")) +
ylab(paste0("PC2: ", pca_percentvar[2], "% variance")) +
geom_text_repel(aes(label=samp), show.legend=F) +
theme_classic() +
theme(legend.position="top", legend.title=element_blank())
p
ggsave(p, filename=paste0(output_prefix,".DESeq2.normalized.rlog.pca.pdf"),width=13.5,height=15,units=c("cm"))
pca_percentvar <- data.frame(PC=colnames(pca_x), Variance=pca_percentvar)
write.table(pca_percentvar, file=paste0(ehbio_output_prefix,".DESeq2.pca_pc_weights.xls"), sep="\t", quote=F, row.names=F, col.names=T)
下图是 pca_individual 的数据结构:
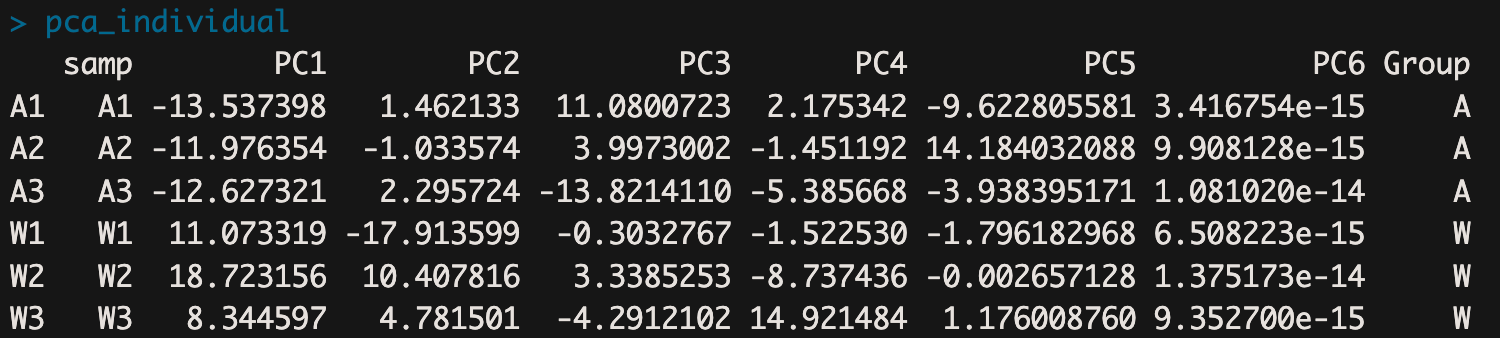
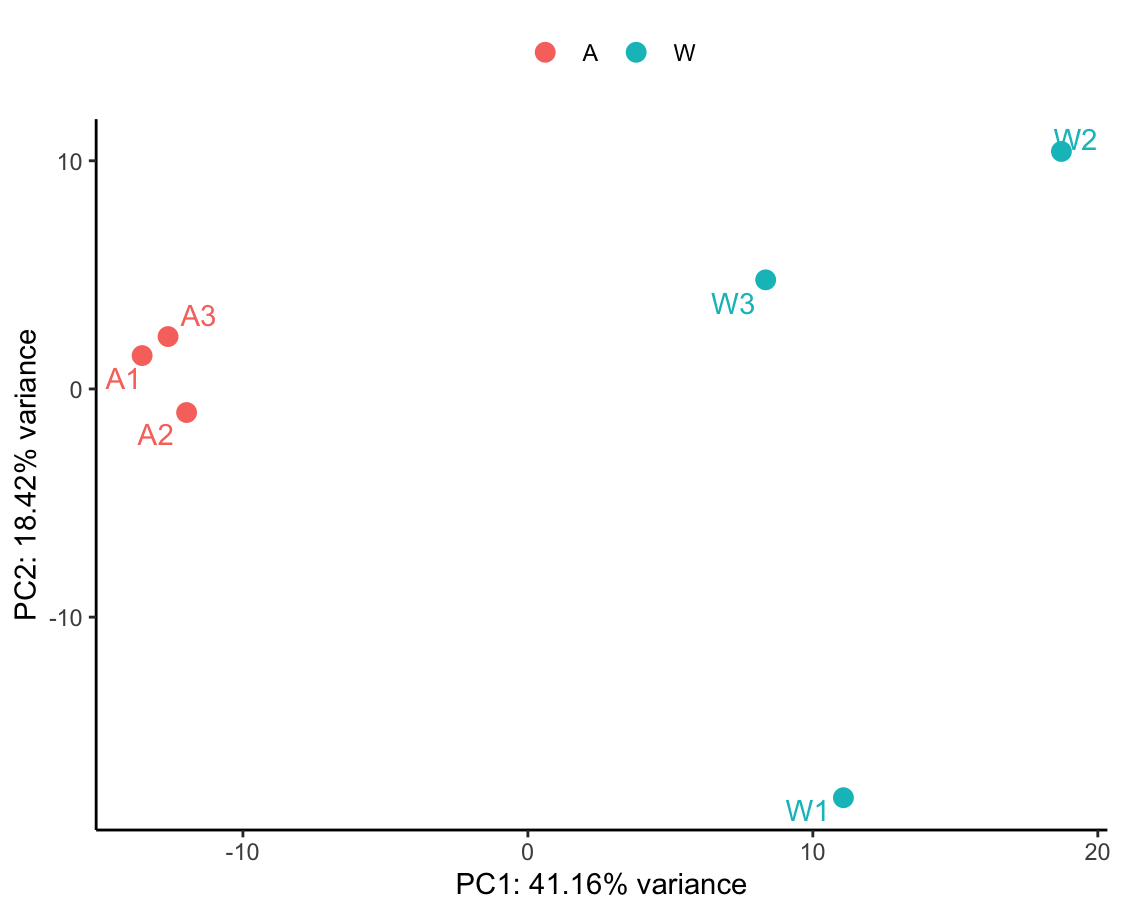
我们选择了 PC1 和 PC2 进行画图
PC1-PC6,为什么只选择了PC1和PC2进行分析,因为PC1和PC2是绘图时最常用的,它们通常解释了最大的方差。
这一步得到的PCA图:
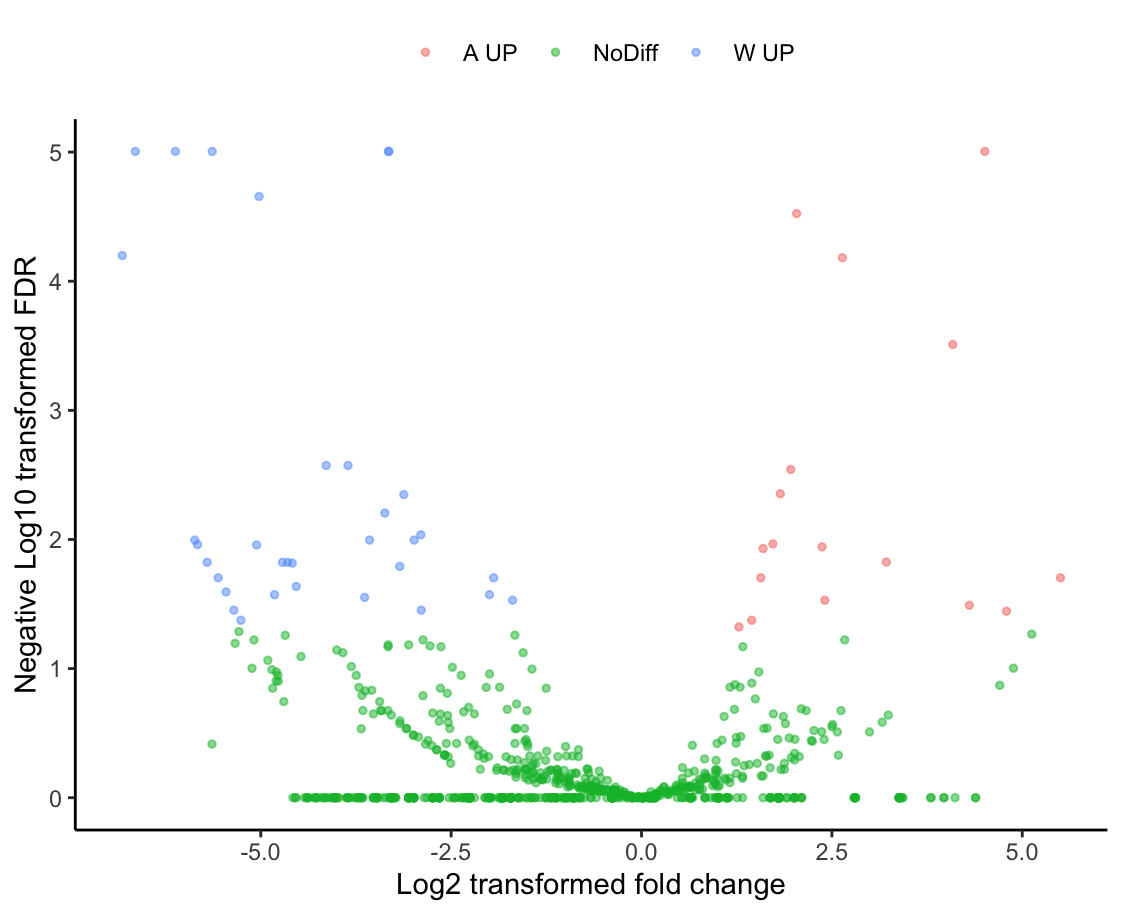
- 火山图
## 两组样品基因差异分析
##################################
## 火山图
##################################
if (file.exists(paste0(output_prefix,".DESeq2.all.DE"))) file.remove(paste0(output_prefix,".DESeq2.all.DE"))
# 注意这两个样本名字需要自己定义,两个样本的比较
# 定义待比较的两个样本组名
sampleA <- "A"
sampleB <- "W"
# 后面无需修改
print(paste("DE genes between", sampleA, sampleB, sep=" "))
# 设定差异表达分析的对比条件
contrastV <- c("Group", sampleA, sampleB)
# 使用DESeq2的results函数,基于给定的对比条件进行差异表达分析。
# contrast=contrastV 因为需要比较两个组的差异表达,需要使用 contrast 参数来明确指定需要比较的组
res <- results(dds, contrast=contrastV)
# 获取 sampleA 的正常化基因计数
baseA <- counts(dds, normalized=TRUE)[, colData(dds)$Group == sampleA]
if (is.vector(baseA)){
baseMeanA <- as.data.frame(baseA)
} else {
baseMeanA <- as.data.frame(rowMeans(baseA))
}
baseMeanA <- round(baseMeanA, 3)
colnames(baseMeanA) <- sampleA
# 获取 sampleB 的正常化基因计数
baseB <- counts(dds, normalized=TRUE)[, colData(dds)$Group == sampleB]
if (is.vector(baseB)){
baseMeanB <- as.data.frame(baseB)
} else {
baseMeanB <- as.data.frame(rowMeans(baseB))
}
baseMeanB <- round(baseMeanB, 3)
colnames(baseMeanB) <- sampleB
# 将sampleA和sampleB的数据与 DESeq2的差异表达分析结果合并
res <- cbind(baseMeanA, baseMeanB, as.data.frame(res))
res <- data.frame(ID=rownames(res), res)
# 数据后处理,如p值和padj的调整
## 计算 sampleA 和 sampleB 的标准化计数的平均值,然后保留3位小数并存入res的baseMean列中
res$baseMean <- round(rowMeans(cbind(baseA, baseB)),3)
## 把 res$padj 列中所有的 NA 值替换为 1
res$padj[is.na(res$padj)] <- 1
## 把 res$pvalue 列中所有的 NA 值替换为 1
res$pvalue[is.na(res$pvalue)] <- 1
## log2FoldChange代表的是两组之间的基因表达量的对数2倍数变化,它可以用来表示基因的上调或下调程度。
## 对res数据框中的log2FoldChange列进行四舍五入,取到3位小数。
res$log2FoldChange <- round(res$log2FoldChange,3)
## 使用 formatC 函数进行格式化 padj 和 pvalue 列的值。as.numeric 确保结果是数字类型。
## 这可能是为了确保 padj 值在后续的操作中具有正确的格式和数据类型。
res$padj <- as.numeric(formatC(res$padj))
res$pvalue <- as.numeric(formatC(res$pvalue))
# 根据调整后的p值对结果进行排序
## order(res$padj) 会返回一个整数向量,这个向量代表了 res$padj 的值从小到大的索引顺序。
res <- res[order(res$padj),]
# 为结果设置文件名
comp314 <- paste(sampleA, "_vs_", sampleB, sep=".")
file_base <- paste(output_prefix, "DESeq2", comp314, sep=".")
file_base1 <- paste(file_base, "results.xls", sep=".")
# 保存完整的差异表达分析结果为xls文件
res_output <- as.data.frame(subset(res,select=c('ID',sampleA,sampleB,"baseMean",'log2FoldChange','pvalue', 'padj')))
write.table(res_output, file=file_base1, sep="\t", quote=F, row.names=F)
# 提取并保存上调的差异表达基因为xls文件
res_de <- subset(res, res$padj<0.05, select=c('ID', sampleA,
sampleB, 'log2FoldChange', 'padj'))
res_de_up <- subset(res_de, res_de$log2FoldChange>=1)
file <- paste(output_prefix, "DESeq2",sampleA, "_higherThan_", sampleB, 'xls', sep=".")
write.table(as.data.frame(res_de_up), file=file, sep="\t", quote=F, row.names=F)
res_de_up_id <- subset(res_de_up, select=c("ID"))
#file <- paste(file_base, "DE_up_id", sep=".")
file <- paste(output_prefix, "DESeq2",sampleA, "_higherThan_", sampleB,'id.xls', sep=".")
write.table(as.data.frame(res_de_up_id), file=file, sep="\t",
quote=F, row.names=F, col.names=F)
# 如果上调基因的数量大于0,则追加到总的差异表达基因文件中
if(dim(res_de_up_id)[1]>0) {
res_de_up_id_l <- cbind(res_de_up_id, paste(sampleA, "_higherThan_",sampleB, sep="."))
write.table(as.data.frame(res_de_up_id_l), file=paste0(output_prefix,".DESeq2.all.DE"),
sep="\t",quote=F, row.names=F, col.names=F, append=T)
}
# 提取并保存下调的差异表达基因为xls文件
res_de_dw <- subset(res_de, res_de$log2FoldChange<=(-1)*1)
file <- paste(output_prefix, "DESeq2",sampleA, "_lowerThan_", sampleB, 'xls', sep=".")
write.table(as.data.frame(res_de_dw), file=file, sep="\t", quote=F, row.names=F)
res_de_dw_id <- subset(res_de_dw, select=c("ID"))
file <- paste(output_prefix, "DESeq2",sampleA, "_lowerThan_", sampleB, 'id.xls', sep=".")
write.table(as.data.frame(res_de_dw_id), file=file, sep="\t",
quote=F, row.names=F, col.names=F)
if(dim(res_de_dw_id)[1]>0) {
res_de_dw_id_l <- cbind(res_de_dw_id, paste(sampleA, "_lowerThan_",sampleB, sep="."))
write.table(as.data.frame(res_de_dw_id_l), file=paste0(output_prefix,".DESeq2.all.DE"),
sep="\t",quote=F, row.names=F, col.names=F, append=T)
}
# 在res_output数据框中为每个基因添加一个分类标签 'level'
# 如果调整后的p值 <= 0.05 且 log2FoldChange >= 1,将其标记为sampleA上调
# 如果调整后的p值 <= 0.05 且 log2FoldChange <= -1,将其标记为sampleB上调
# 其他的所有情况(不显著或log2FoldChange在-1到1之间的)都标记为 'NoDiff'
res_output$level <- ifelse(res_output$padj<=0.05, ifelse(res_output$log2FoldChange>=1, paste(sampleA,"UP"), ifelse(res_output$log2FoldChange<=1*(-1), paste(sampleB,"UP"), "NoDiff")) , "NoDiff")
# 转换调整后的p值为其负对数10转换。这是为了在火山图上展示更好的视觉效果。
res_output$padj <- (-1) * log10(res_output$padj)
# 如果负对数10转换后的调整p值大于5,则将其替换为5.005(这可能是为了避免图上有极端的点)
res_output$padj <- replace(res_output$pad, res_output$pad>5, 5.005)
# 计算火山图x轴的范围。这里的目的是找出log2FoldChange的绝对值的最大值,
# 然后对其进行上取整,使得火山图在x轴上左右对称。
boundary = ceiling(max(abs(res_output$log2FoldChange)))
#
boundary = ceiling(max(abs(res_output$log2FoldChange)))
# 火山图
# 使用ggplot2包创建火山图
# x轴表示log2转换后的倍数变化,y轴表示调整后的p值的负对数10转换
# 点的颜色基于之前定义的'level'
p = ggplot(res_output, aes(x=log2FoldChange,y=padj,color=level)) +
#geom_point(aes(size=baseMean), alpha=0.5) + theme_classic() +
geom_point(size=1, alpha=0.5) +
theme_classic() +
xlab("Log2 transformed fold change") + ylab("Negative Log10 transformed FDR") +
# 设置x轴范围。这里根据之前计算的boundary来确定x轴的范围,使火山图左右对称。
xlim(-1 * boundary, boundary) +
theme(legend.position="top", legend.title=element_blank())
ggsave(p, filename=paste0(file_base1,".volcano.pdf"),width=13.5,height=15,units=c("cm"))
p
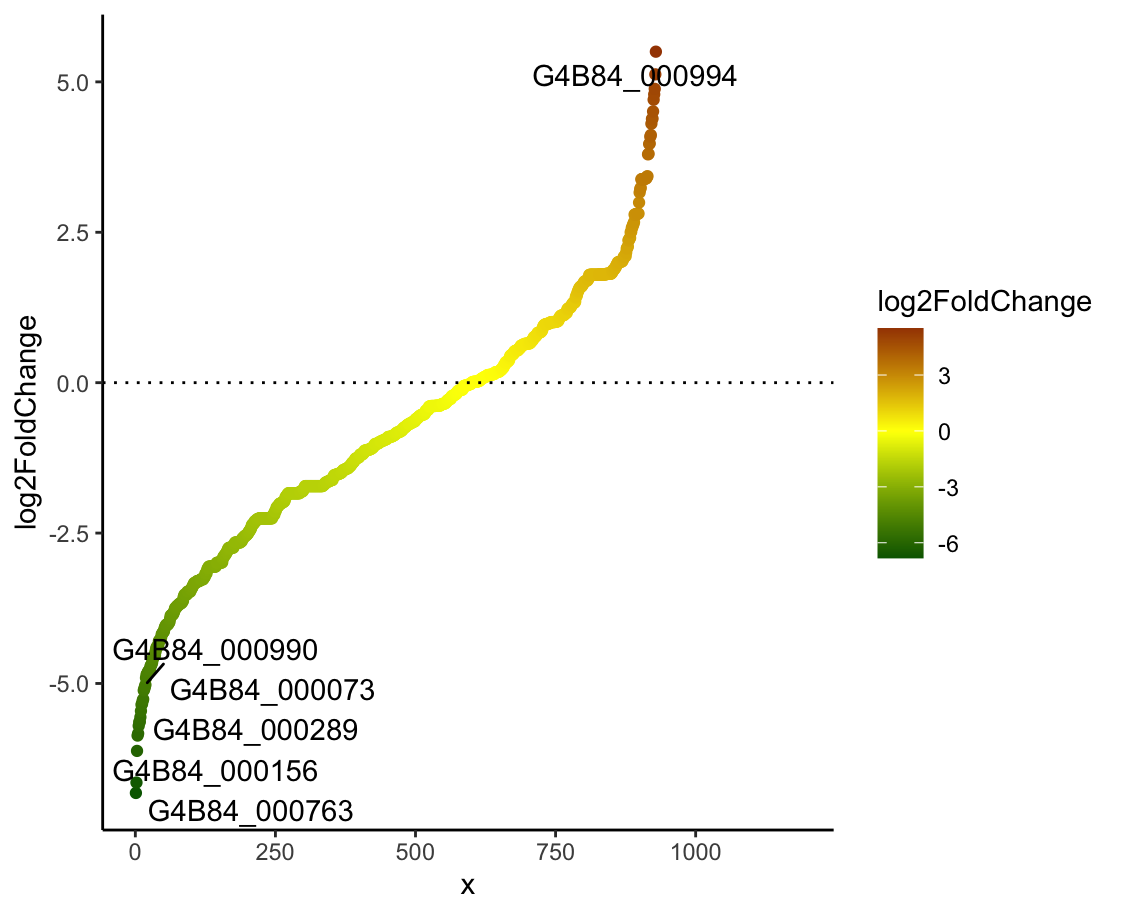
- 不知道啥图
## 根据log2FolcChange排序基因
##################################
## 不知道啥图
##################################
#head(res_output)
# 整理数据,按差异倍数排序,增加横轴,选择log2差异倍数大于5的标记基因名字
res_output_line <- res_output[order(res_output$log2FoldChange),]
res_output_line$x <- 1:nrow(res_output_line)
res_output_line[abs(res_output_line$log2FoldChange)<5 | res_output_line$level=="NoDiff", "ID"] <- NA
head(res_output_line)
library(ggrepel)
p <- ggplot(res_output_line, aes(x=x, y=log2FoldChange)) + geom_point(aes(color=log2FoldChange)) +
scale_color_gradient2(low="dark green", mid="yellow", high= "dark red", midpoint = 0) +
theme_classic() + geom_hline(yintercept = 0, linetype="dotted")
p
# 标记基因名字
p1 = p + geom_text_repel(aes(label=ID))
ggsave(p1, filename=paste0(output_prefix,".log2FoldChange.pdf"),width=13.5,height=15,units=c("cm"))
- Top20差异显著基因热图
## Top20差异显著的基因
##################################
## HeatMap图
##################################
res_de_up_top20_id <- as.vector(head(res_de_up$ID,20))
res_de_dw_top20_id <- as.vector(head(res_de_dw$ID,20))
res_de_top20 <- c(res_de_up_top20_id, res_de_dw_top20_id)
res_de_top20
res_de_top20_expr <- normalized_counts[res_de_top20,]
res_de_top20_expr
library(pheatmap)
p = pheatmap(res_de_top20_expr, cluster_row=T, scale="row", annotation_col=sample)
p
ggsave("heatmap.pdf", plot=p, device = 'pdf')
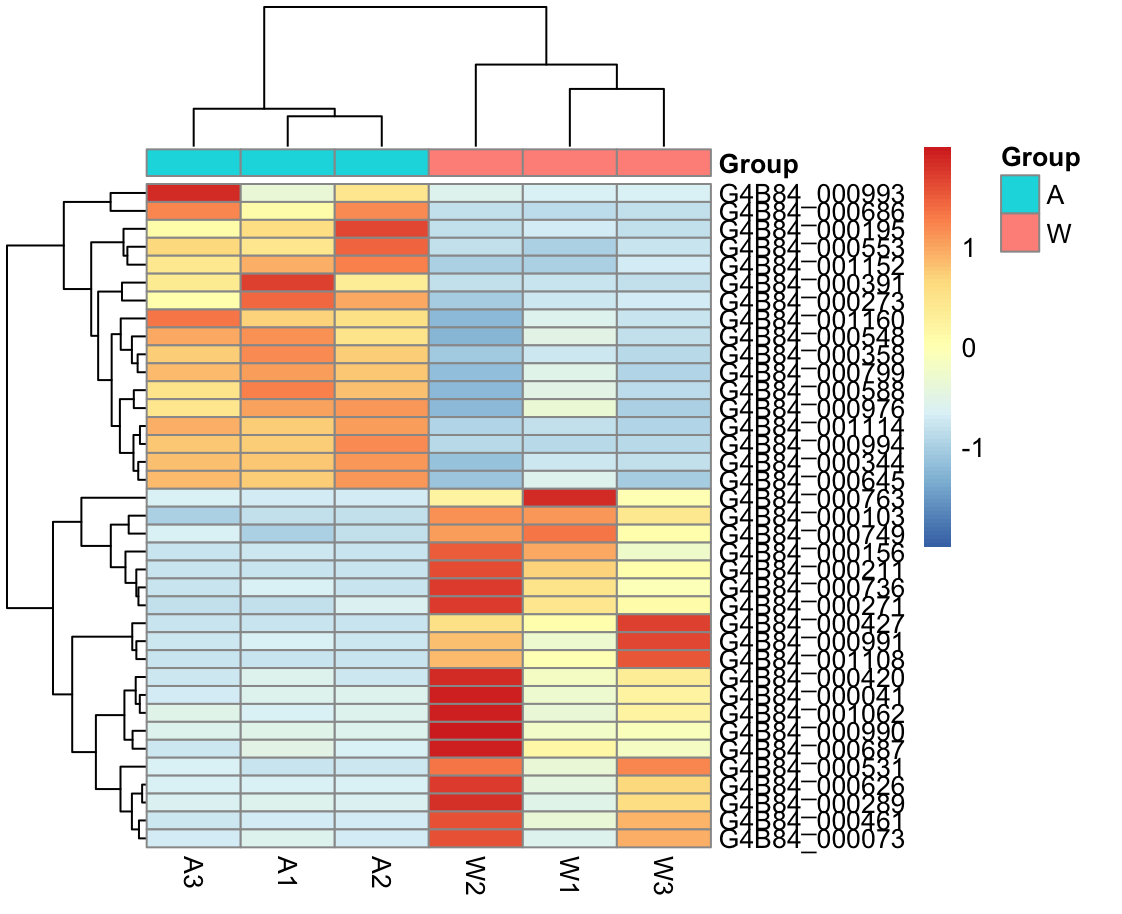
- 差异基因散点图
## 差异基因散点图
##################################
# 增加基因名字列
res_de_top20_expr2 <- data.frame(Gene=rownames(res_de_top20_expr), res_de_top20_expr)
head(res_de_top20_expr2)
# 转换为长格式
res_de_top20_expr2 <- melt(res_de_top20_expr, id=c("Gene"))
colnames(res_de_top20_expr2) <- c("Gene", "Sample", "Expression")
head(res_de_top20_expr2)
# 图形绘制
p = ggplot(res_de_top20_expr2, aes(x=Gene, y=Expression)) + geom_point(aes(color=Sample), alpha=0.5) + theme_classic() +
theme(axis.text.x = element_text(angle = 90, vjust = 1, hjust = 1),
axis.title.x = element_blank()) + ylab("Normalized expression value") + scale_y_log10()
p
ggsave(p, filename=paste0(output_prefix,".expression.point.pdf"),width=13.5,height=15,units=c("cm"))
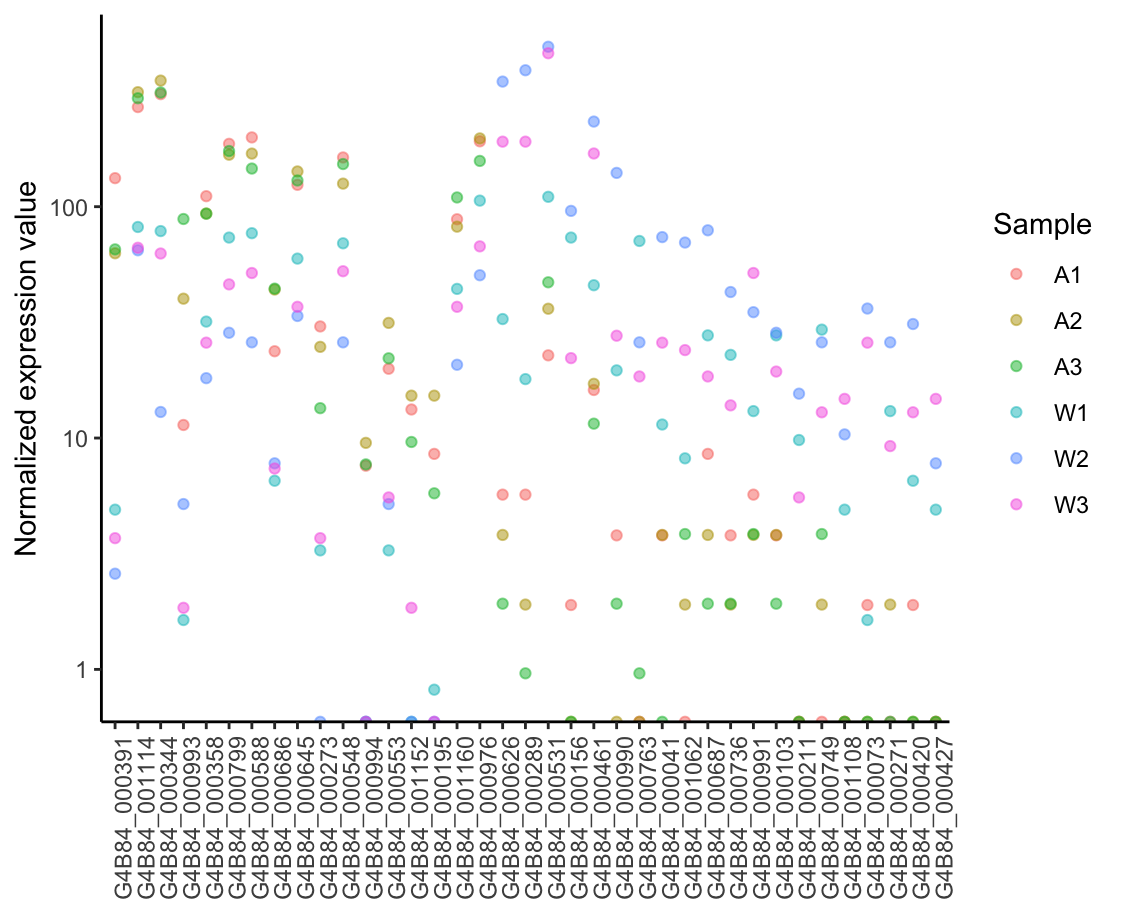
以上就是本流程的DESeq分析。下面的功能富集分析在下一篇文章中详述。
功能富集分析
- GO 富集分析
- KEGG通路分析
- GSEA分析