全文链接:http://tecdat.cn/?p=30051
原文出处:拓端数据部落公众号
蛋白质组学
•研究生物体产生的全部蛋白质。
• Foci:鉴定、结构测定、生物标志物、通路、表达。
基质辅助激光解吸飞行时间质谱(MALDI-TOF)法示例频谱
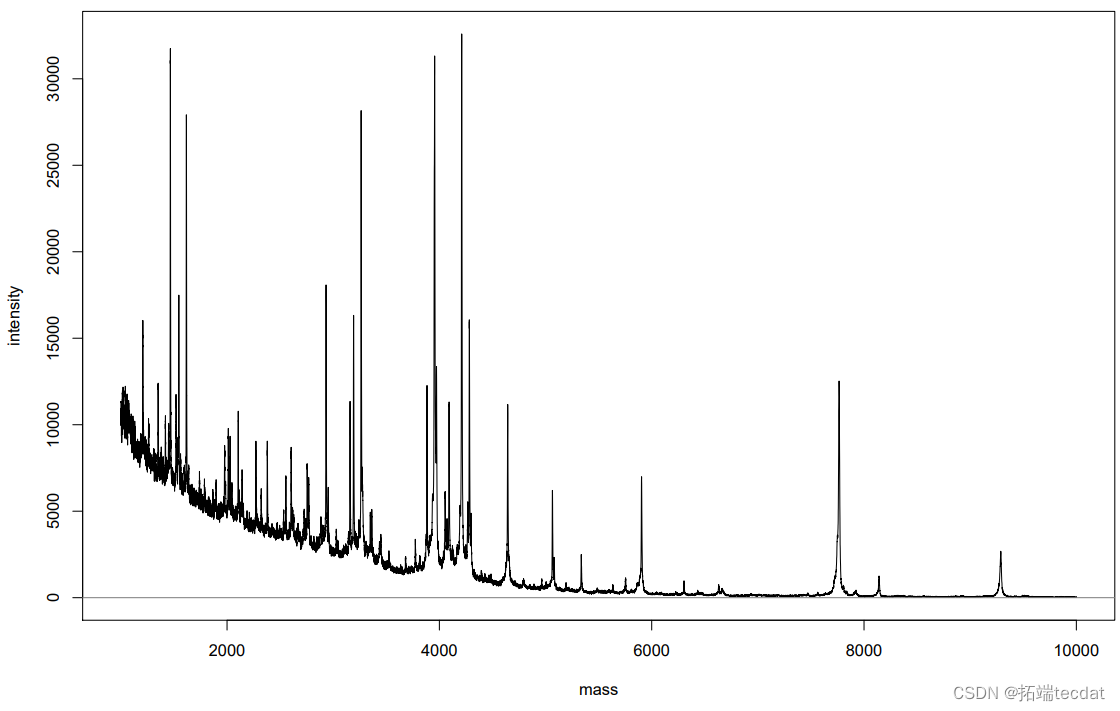
研究动机
只有相对较少的开源软件解决方案可用,而R平台则很少。
• 处理技术和生物复制的必要性。
• 相对强度的定量令人不满意
• 研究光谱校准对临床预后的影响。
• 模块化且易于定制的分析程序
示例数据
血清肽多姆分析显示血小板因子 4 是与胰腺癌相关的潜在鉴别肽
G.M. Fiedler, A.B. Leichtle, J. Kase et al Clin Cancer Res June 1, 2009 15:3812-3819
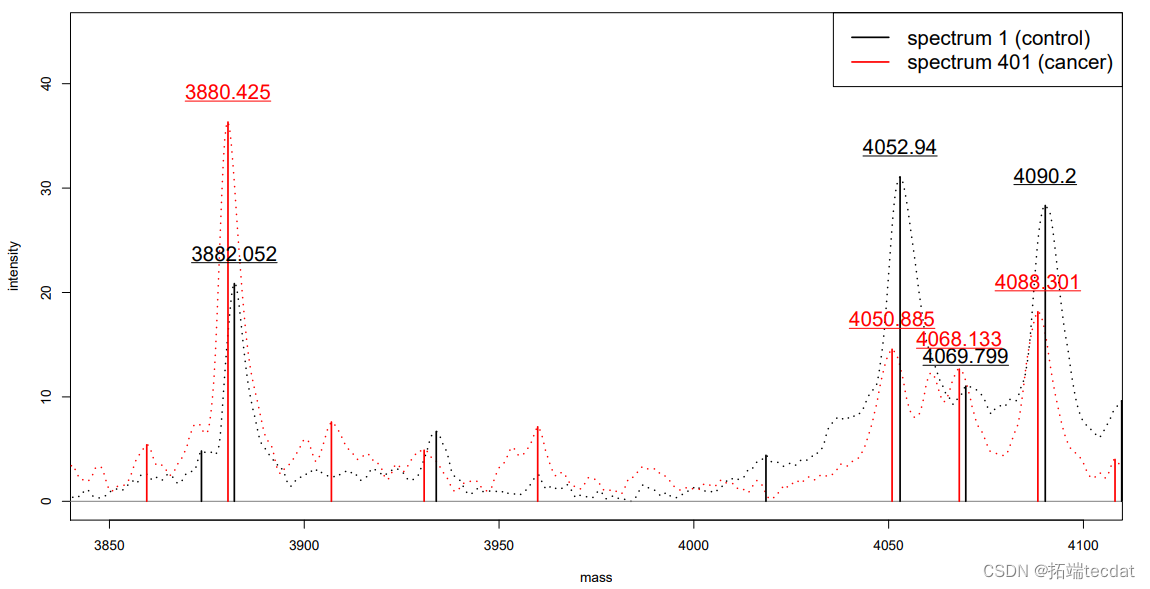
“两个显著峰(m/z 3884;5959)对区分患者和健康对照的敏感性为86.3%,特异性为97.6%。"
“基于MALDI-TOOF MS的血清肽球分析允许发现和验证血小板因子4 [m / z 3884,7767;S.G.]作为胰腺癌的新鉴别标志物。
动手操作:文件导入
- > spectra[[1]]

动手操作:画图
> abline(v=3884, col="blue")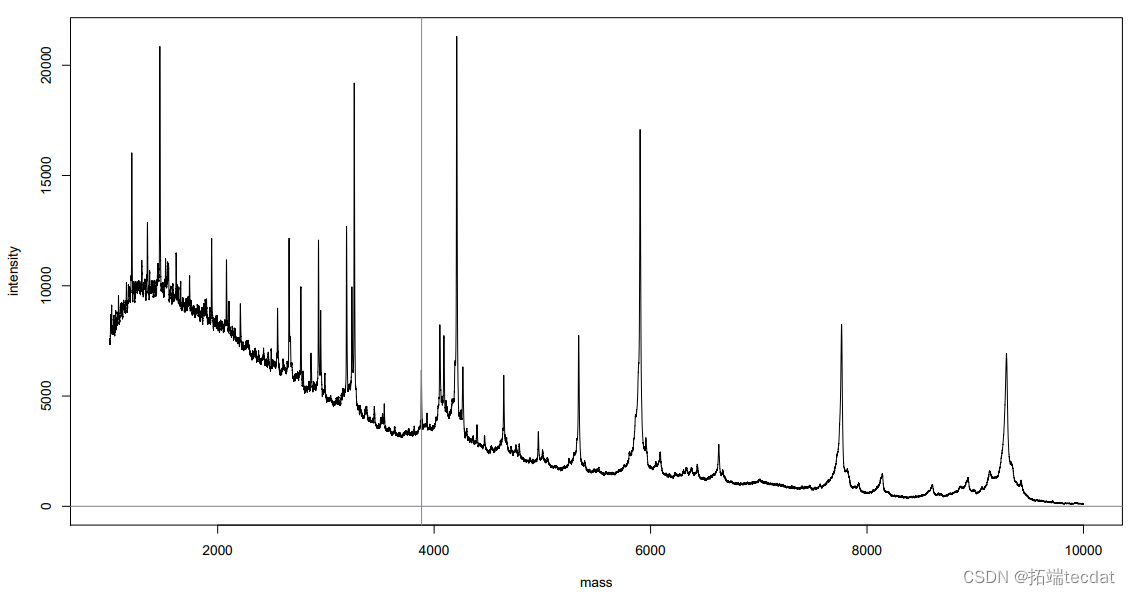
动手操作:画图m/z 3884
> plot(spectra[[1]] 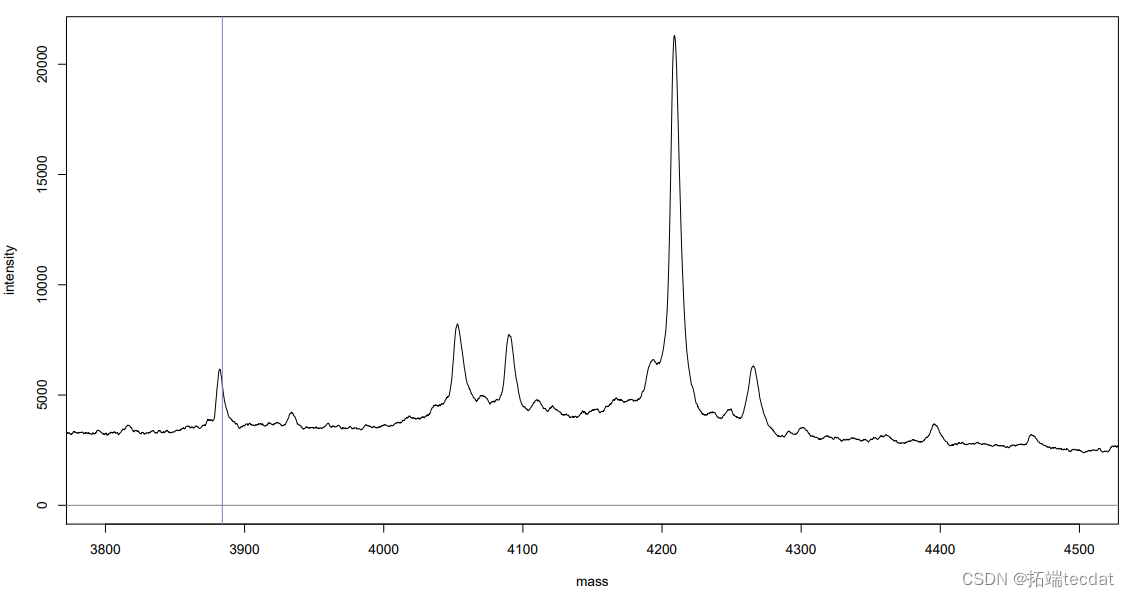
动手操作:方差稳定和平滑
- > spea <- lapply(sptra, transfensty, fun=moAvg)
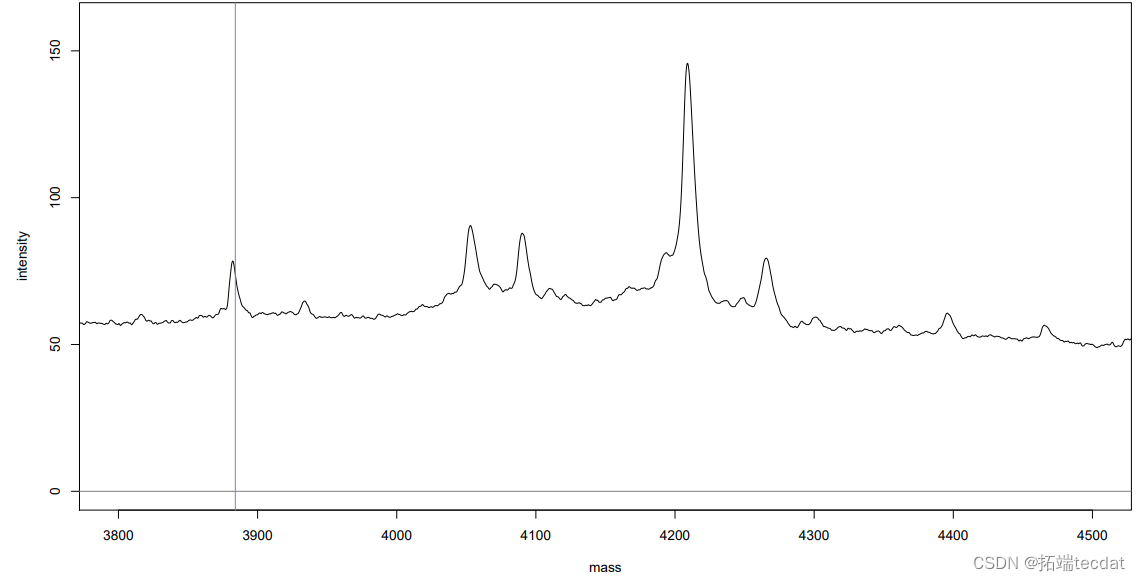
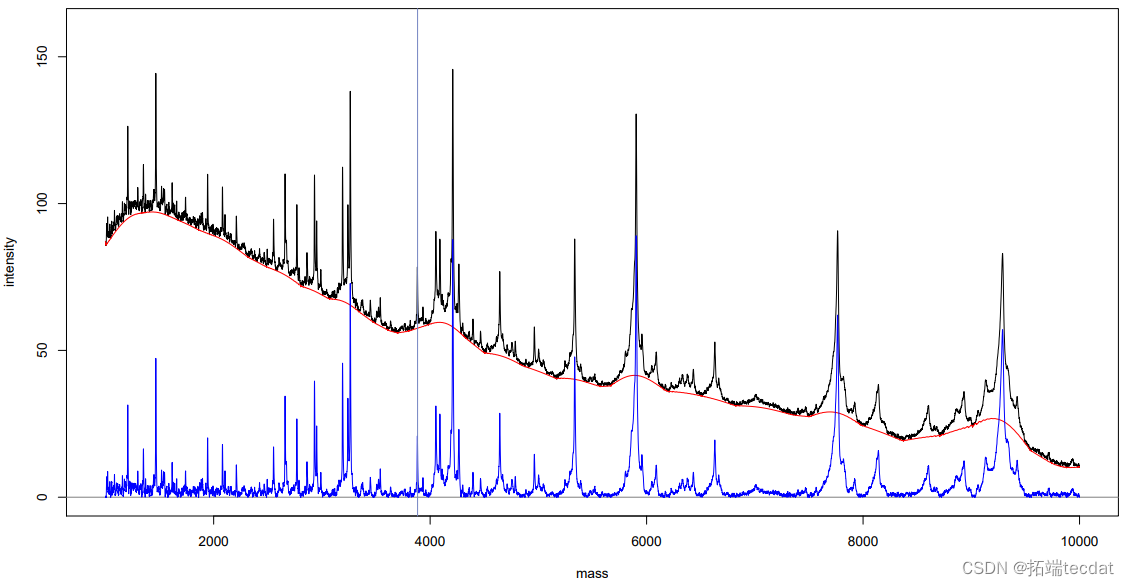
动手实践:基线校正
- > plot(spea[[1]]
- > lines(b, col="red");
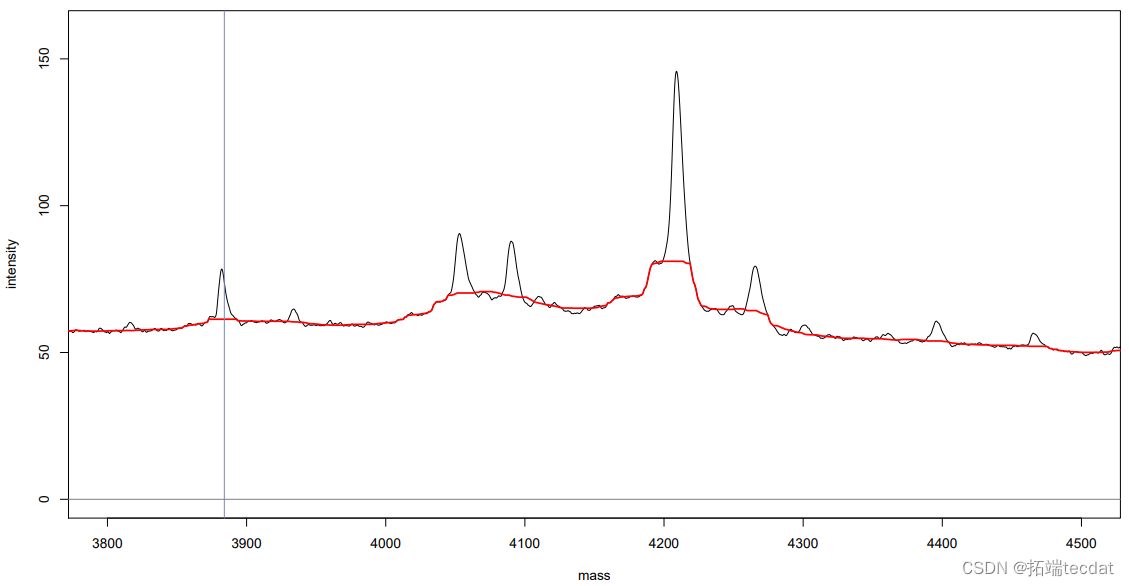
动手操作:基线校正 – SNIP
> bl <- estiline(specta[[1]]
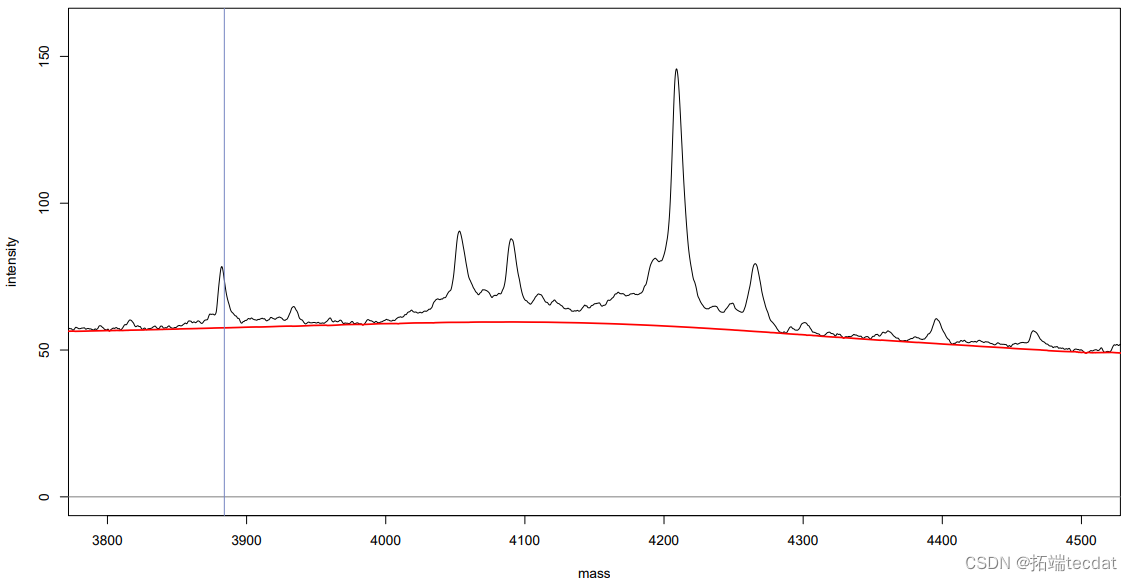
动手操作:基线校正 – SNIP
- > speca <- lapply(spe, remoeline)
- > lines(specra[[1]])
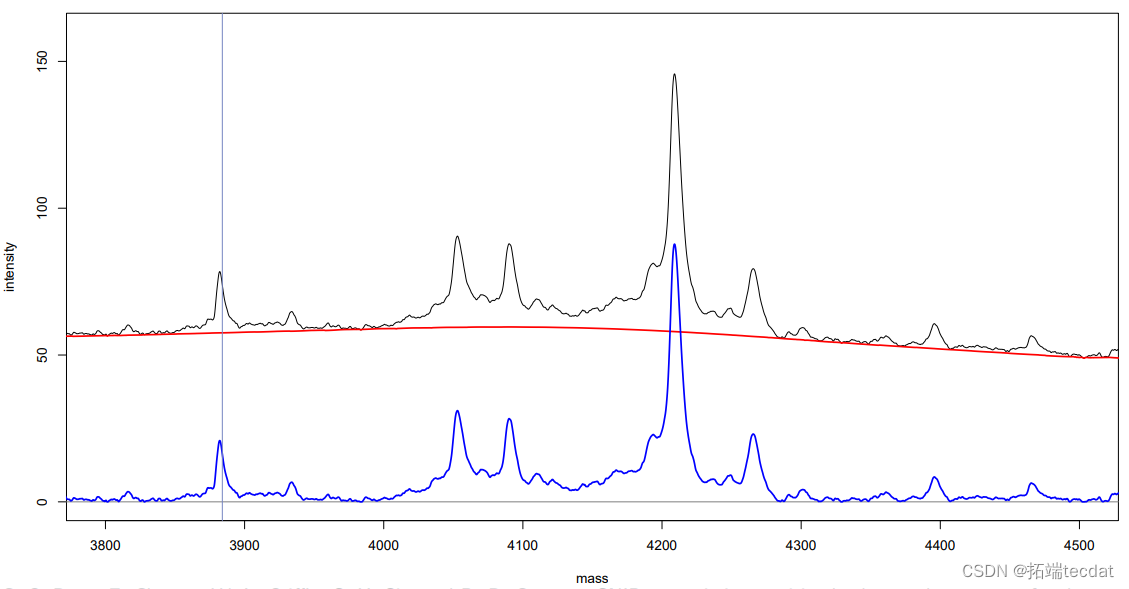
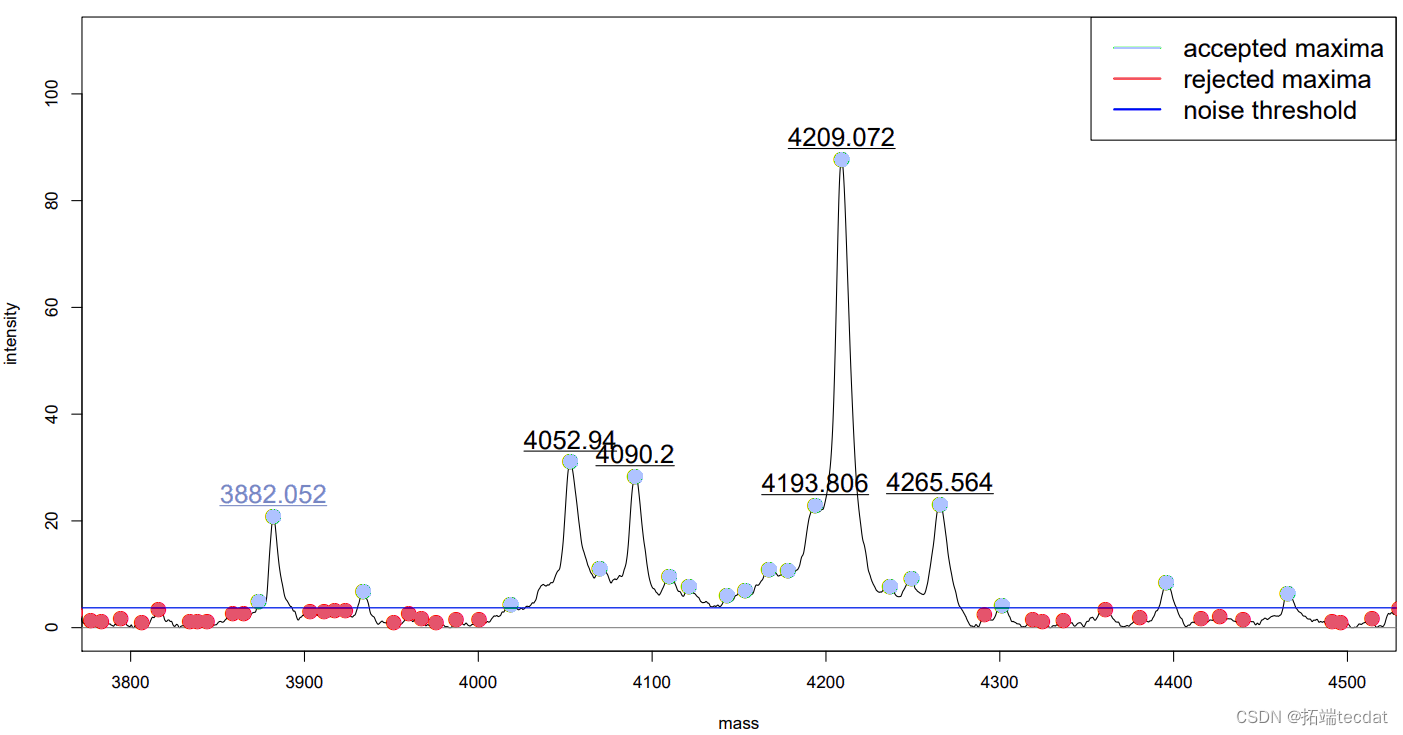
实践:峰值检测

> peas[[1]]

- > top <- intnsity(p) %in% sort(int
- > labePeak
单光谱工作流程
- > peaks <- lapply(spetra, detecPeaks)
多光谱比较

最受欢迎的见解
1.R语言分布式滞后非线性模型(DLNM)空气污染研究温度对死亡率影响建模
2.R语言分布滞后线性和非线性模型(DLNM)分析空气污染(臭氧)、温度对死亡率时间序列数据
3.R语言群组变量选择、组惩罚GROUP LASSO套索模型预测分析新生儿出生体重风险因素数据和交叉验证、可视化
4.R语言逻辑回归、随机森林、SVM支持向量机预测FRAMINGHAM心脏病风险和模型诊断可视化
5.R语言非线性混合效应 NLME模型(固定效应&随机效应)对抗哮喘药物茶碱动力学研究
6.R语言使用限制平均生存时间RMST比较两条生存曲线分析肝硬化患者
7.分类回归决策树交互式修剪和更美观地可视化分析细胞图像分割数据集
8.PYTHON深度学习实现自编码器AUTOENCODER神经网络异常检测心电图ECG时间序列
9.R语言如何在生存分析与Cox回归中计算IDI,NRI指标
标签:分析,3884,峰值检测,语言,组学,光谱,动手,MALDI,TOF From: https://www.cnblogs.com/tecdat/p/16866418.html